Kamagra enthält Sildenafilcitrat als pharmakologisch aktiven Bestandteil. Dieser hemmt selektiv die Phosphodiesterase-5 und erhöht dadurch die Konzentration von cGMP im Corpus cavernosum. Der Effekt ist zeitlich begrenzt, da die Halbwertszeit von Sildenafil etwa vier Stunden beträgt. In der galenischen Form als Mundgel erfolgt die Resorption besonders rasch, was zu einem schnelleren Wirkeintritt führt. Der Abbau erfolgt überwiegend hepatisch über CYP3A4, wobei ein aktiver Metabolit entsteht, der zur Gesamtwirkung beiträgt. Typische Nebenwirkungen ergeben sich aus der Vasodilatation, darunter leichte Kopfschmerzen und nasale Kongestion. In klinischen Beschreibungen wird kamagra oral jelly im Zusammenhang mit der schnelleren Absorption erwähnt.
022478_cloetta_bro_ug
OF ATHEROSCLEROSIS AND A POTENTIAL TARGET
Atherosclerosis, and its devastating complications cerebral andmyocardial infarction and gangrene of the extremities, is the leadingcause of death. Atherogenesis in humans develops over many years,often reckoned in decades. Early lesion formation may even occur inadolescence. Lesion progression depends on genetic make-up, genderand certain well-recognized risk factor as well as a number of non-traditional risk factor that are currently the subject of intense investi-gation.
Our concepts of atherogenesis have evolved from vague ideas of inevit-able degeneration to a much better defined scenario of molecular andcellular events. As we enhance our understanding of its fundamentalmechanism, we can begin to approach atherogenesis as a modifiablerather than ineluctable process. Recently, inflammatory and immunolo-gical mechanisms have been increasingly implicated in the pathogene-sis of this disease. Postulates for the involvement of innate and adaptiveimmunity with cellular as well as humoral components have met withconsiderable experimental support. It is now well recognized that atherosclerotic lesions contain activated, immunocompetent cells,including T lymphocytes and monocyte/macrophages. Inflammatoryand/or immune mechanisms appear to be particularly active whenplaques are activated and rupture and they may therefore cause acutecoronary syndromes or stroke. Increasing knowledge of the basicmechanisms enables us to understand how current therapies for ather-osclerosis may act. Insights derived from recent scientific advancesshould aid the discovery of new therapeutic targets that would stimul-ate development of novel treatment. Such new treatments could furtherreduce the considerable burden of morbidity and mortality due to this
modern scourge, and reduce reliance on costly technologies thataddress the symptoms rather than the cause of atherosclerosis. Eventually, mastery of the cell and molecular biology of atherosclero-sis may permit development of novel strategies for mitigating this pre-valent disease. Our research group has produced several evidences toshown new mechanism of immune cell cross-talk and activation. In thisreview I will describe some anti-inflammatory agents capable reducingthese inflammatory processes and atherosclerosis.
Health trends in the next 25 years will be determined by the ageing ofthe world’s population, the decline in age-specific mortality rates fromcommunicable, maternal, perinatal, and nutritional disorders, the spreadof HIV, and the increase in tobacco-related mortality and disability. Today, ischemic heart disease is the leading cause of death in develo-ped countries with more than 3 million death/year, followed by stroke(1.6 million) and lung cancer (0.7 million) [1]. By 2020, based oncurrent trends, ischemic disease will be the leading cause of diseaseburden worldwide, followed by unipolar major depression, road trafficaccidents, stroke and chronic obstructive lung disease. For worldwideischeamic disease only, the number of Disability-Adjusted Life Years orDALYs estimated for 2020 is about 82.3 millions per year [2]. Thus,despite changes in lifestyle and the use of new pharmacological appro-aches, atherosclerosis and its devastating clinical complications such asischemia and infarction of the heart, the brain and other vital organs,ruptured aortic aneurysm and peripheral vascular insufficiency, con-tinue to account for the majority of mortality and morbidity in the adultpopulation of industrialized countries, and would be so worldwide by2020 [3].
Atherosclerosis is a progressive disease process that generally begins inchildhood and has clinical manifestations in middle to late adulthood. Atherosclerosis focally affects the aorta, carotid, coronary, iliac andfemoral arteries. This disease is characterized by lesions (called ather-oma), localized in the intima of the vessel wall. From the initial phases
of leukocyte recruitment, to eventual rupture of the vulnerable atheros-clerotic plaque, inflammatory mediators appear to play a key role in the pathogenesis of atherosclerosis [4]. Thus, atherosclerosis is not anylonger considered to be a degenerative process because of the accumul-ation of lipid and necrotic debris in the advanced lesions. Indeed, alreadyin 1856 Virchow proposed that “A form of low-grade injury to theartery wall resulted in a type of inflammatory insudation, which in turn caused increased passage and accumulation of plasma consti-tuents in the intima of the artery”[5]. Three fundamental biologicalprocesses generate atherosclerotic lesions: 1) LDL-cholesterol infil-tration and activation within the intima of arteries; 2) endothelial acti-vation and dysfunction, with adhesion molecule expression; 3) leuko-cyte recruitment from the blood stream with the consequent productionof pro-inflammatory cytokines, chemokines and proteases, responsiblefor the maintaining of these inflammatory process within the vesselwall. These soluble mediators induce also the proliferation on smoothmuscle cells and the production of acute phase reactants by the liver [6]. This complex process induces the developing of a systemic pro-in-flammatory state in which different soluble factors and cellular playersare involved. Accumulating evidence suggests that inflammatory pro-cesses play a fundamental role in each of these stages in atherogenesis. Although the first event of atherogenetic cascade remains unknown, theinflammatory processes have to be considered as key components ofthe developing of the disease and promising targets for novel and moreselective treatments. Therefore, the discovery of new molecularmechan-isms, which could be contrasted by novel therapeutic strate-gies, represents an important possibility for humans to mitigate athe-rosclerosis. In this regard, the development of murine models of athe-rosclerosis has revolutionized the approach to evaluating potential roles of specific factors in lesion development. These models are basedon targeted disruption of the low-density lipoprotein (LDL) receptor or apolipoprotein E (apoE) genes [7]. Feeding of these knock out micewith cholesterolrich diet results in massive hypercholesterolemia and, thus, to atherosclerosis development. The implication of variousinflammatory mediators in atherosclerosis has been clarified by cross-ing these animals with mice that overexpress or lack possible candidategenes.
My main research field was focused on leukocyte recruitment to in-flammatory sites and inflammatory cell activation. Then, I identifiedpromising therapeutic strategies (statins, anti-chemokines and cannabi-noids) to reduce these inflammatory processes and thus the progressionof atherosclerosis.
Inflammation is a protective reaction against a variety of exogenous(microbial, chemical, physical) or endogenous (immunological, neuro-logical) disturbances, which is characterized by the accumulation ofspecific subsets of leukocytes to sites of infection or tissue damage, andtheir subsequent activation [8]. The attraction of leukocytes to tissues isessential for inflammation and the host response to infection [9]. Thismigration is a directional, nonrandom and selective process. The pro-cess of leukocyte trafficking manifests itself as inflammation with fourclassic cardinal signs: redness, swelling, heat and pain. In order torecruit leukocytes, the capillary blood flow and the vascular permeabi-lity are increased. This allows for enhanced migration of the leukocytesthrough the vascular endothelium, which is the boundary between thecapillaries and the tissue, towards the site of inflammation. Dependingon the cause, inflammation can resolve rapidly or develop into a com-plex process involving different leukocytes as well as endothelial andmesenchymal cells. When an infection or a lesion appears, the organ-ism acts as quickly as possible in order to get rid of the injury. Amongthe first immune cells to arrive at the lesional site are neutrophils, whichinitiate a rapid, nonspecific phagocytic response [10, 11]. These cellsproduce toxic substances including proteases and oxygenic radicals that suppress the pathogen quickly but non-specifically. Whilst this pro-cess is efficient, a more specific antigenic recognition mechanism hasevolved. In the specific antigen recognition process, antigen-presentingcells migrate to the site where antigen is present, followed by specificsubsets of T and B lymphocytes [12]. The combination and activationof leukocytes generates the antigen-specific immune response, whichresults in the production of appropriate antibodies and activation ofcytotoxic T lymphocytes [8].
There are two classes of inflammation: acute inflammation, which is ofshort duration and is characteristically accompanied by plasma fluidexudates and neutrophils accumulation; and chronic inflammationwhich involves other leukocyte types such as monocytes/macrophages,T cells, eosinophils, basophils, mast cells and dendritic cells. This classof inflammation is of longer duration and is characterized by densecellular infiltrates. Emerging evidence supports involvement an im-plication of chronic inflammation as the crucial cornerstone of ather-ogenesis [13–17].
A role for inflammation has become well established over the pastdecade or more in theories describing the atherosclerotic disease pro-cess. From a pathological viewpoint, all stages, ie, initiation, growth,and complication of the atherosclerotic plaque might be considered tobe an inflammatory response to injury. In a variety of animal models of atherosclerosis, signs of inflammation occur hand-in-hand withincipient lipid accumulation in the artery wall. For example, bloodleukocytes, mediators of host defenses and inflammation, localize inthe earliest lesions of atherosclerosis, not only in experimental animalsbut in humans as well. The basic science of inflammation biologyapplied to atherosclerosis has afforded considerable new insight intothe mechanisms underlying this recruitment of leukocytes. The normalendothelium does not in general support binding of white blood cells. However, early after initiation of an atherogenic diet, arterial endoth-elial cells begin to express on their surface selective adhesion moleculesthat bind to various classes of leukocytes. Interestingly, the foci of in-creased adhesion molecule expression overlap with sites in the arterialtree particularly prone to develop atheroma. Considerable evidencesuggests that impaired endogenous atheroprotective mechanisms occurat branch points in arteries, where the endothelial cells experiencedisturbed flow. For example, absence of normal laminar shear stressmay reduce local production of endothelium-derived NO. This end-ogenous vasodilator molecule also has anti-inflammatory properties[18]. In addition to inhibiting natural protective mechanisms, disturbed
flow can augment the production of certain leukocyte adhesionmolecules [19]. Augmented wall stresses may also promote the pro-duction by arterial smooth muscle cells of proteoglycans that can bind and retain lipoprotein particles, facilitating their oxidative modi-fication and thus promoting an inflammatory response at sites of lesionformation.
Once adherent to the endothelium, monocytes and T lymphocytespenetrate into the intima. Chemoattractant molecules appear to beresponsible for the direct migration of monocytes and T lymphocytesinto the intima at sites of lesion formation [20–23]. Once resident in the arterial wall, the blood-derived inflammatory cells participate inand perpetuate a local inflammatory response. The macrophagesexpress scavenger receptors for modified lipoproteins, permittingthem to ingest lipid and become foam cells. Several pro-inflammatorymediators, such as macrophage colony-stimulating factor (M-CSF)contribute to the differentiation of circulating monocytes into themacrophage foam cell [24]. T lymphocytes likewise encounter signalsthat cause them to elaborate inflammatory cytokines, such as inter-feron-γ, interleukins, or tumor necrosis facto-α, which in turn can stim-ulate macrophages as well as vascular endothelial cells and SMCs [25]. Inflammatory processes not only promote initiation and evolution ofatheroma, but also contribute decisively to precipitating acute throm-botic complications of atheroma. Most coronary arterial thrombi thatcause fatal acute myocardial infarction arise because of a physicaldisruption of the atherosclerotic plaque. The activated macrophageabundant in atheroma can produce proteolytic enzymes capable ofdegrading the collagen that lends strength to the plaque’s protectivefibrous cap, rendering that cap thin, weak, and susceptible to rupture. Interferon arising from the activated T lymphocytes in the plaque canhalt collagen synthesis by SMCs, limiting its capacity to renew thecollagen that reinforces the plaque [8]. Macrophages also producetissue factor, the major procoagulant and trigger to thrombosis found inplaques. Inflammatory mediators regulate tissue factor expression byplaque macrophages, demonstrating an essential link between arterialinflammation and thrombosis [14–16]. Anti-inflammatory effects on atherogenesisCD40
More than a decade ago, discovery of the B lymphocyte receptor CD40,which mediates B cell activation, proliferation and survival whenengaged by antibodies, initiated a search for its respective native ligand. Due to the observation that interaction with T lymphocytes mimics theprocess observed in B lymphocytes after engagement of CD40 by anactivating antibody, several groups independently identified the CD40ligand (also referred to as gp39, TRAP, or TBAM), recently renamedCD154, as the native ligand [26–28]. CD154 was characterized as anintegral membrane protein restricted to activate CD4+ helper T lymph-ocytes. Subsequent studies established that the interaction of CD154with its receptor CD40 is essential for appropriate thymus-dependenthumoral immune response [29–31]. The fundamental role for CD40/CD154 receptor-ligand dyad in these responses was further establishedby the demonstration that mutations in the gene encoding for the ligandresult in the X-linked immunodeficiency hyper IgM syndrome, which ischaracterized by drastic or complete inhibition of the T lymphocyte-helps humoral immune response [32, 33]. Studies of CD40- andCD154-deficient mice further outlined the essential function for thisreceptor-ligand dyad in secondary immune responses. However, it hasbecome evident in recent years that both CD154 and CD40 are express-ed on other leukocytes and, even more interestingly, on non-hemato-poetic cell types as well [34]. The discovery of a broad distributionpattern naturally implied the potential importance of CD40/CD154interactions in processes other than originally consider-ed. Indeed,recent findings revealed elevation of CD40 and CD154 in chronicinflammatory diseases, such as rheumatoid arthritis [35], systemic lupuserythematosus [36], multiple sclerosis [37], and graft-versus-hostdisease [38, 40]. This suggested to us to study the implication of CD40-CD40 ligand interaction in inflammatory processes involving leukocytesand vascular cells and governing atherosclerotic processes. We firstdemonstrated that CD40 and CD154 were highly expressed (most pre-dominantly in the shoulder region of the plaque, the border between thelesion and the unaffected portion of the artery) by atheroma-associatedcells in tissue-section obtained from of human carotid atheroma [41].
Then, we showed that, by inducing the expression of matrix-degradingproteinases and of tissue factor procoagulant, CD40 signalling inmacrophages may contribute to the triggering of acute coronary events[42]. Furthermore, also smooth muscle cells are activ-ated by this systemin an inflammatory microenvironment of atherosclerotic plaques [43]. Also T lymphocytes are activated through CD40 triggering [44]. Themost important evidence of an involvement of this interaction in ather-osclerotic process was strongly support by reduction of atherosclerosisprogression in mice treated with an antibody blocking CD154 signalling[45]. Therefore, we observed that CD40-CD40 ligand interaction iscrucial for several events in atherosclerotic processes.
Chemokines were first identified as a family of chemoattractant cytok-ines, over 10 years ago [46]. Chemokines constitute a large family ofsmall proteins (8-10 kDa) that are involved in both the basal leukocytetrafficking and the activation and recruitment of specific cell popula-tions during disease. Chemokines have a broad spectrum of biologicalactivities [47]. To date, these proteins have been shown to be implicatedin a wide range of immune inflammatory responses:By 2006, the use of either conventional cloning techniques or signalsequence trapping methods brought the number of human chemokinesequences to around 60, most of them identified in past few years. Withthis high number of distinct chemokines known, the extent of complex-ity in the chemokine superfamily is unrivaled within the field of cytok-ines. If the estimate of 100,000 for the total number of distinct humangenes which are transcribed into mRNAs (and ultimately translated intoproteins) is accurate, then it is likely that the entire number of cDNAfragments characterized to date represent probably only 25–35% of thetotal. On this basis, it is possible that the complete number of chemok-ines, when known, could exceed 100 [48].
The chemokines act through receptors belonging to a superfamily thatspan the membrane seven times [49–51]. The number of chemokinereceptors has recently increased significantly. As there are so many
ligands with apparently similar chemoattractant properties, a highlyactive field of research has been to try to identify their respective recep-tors. The explosion in the number of ligands identified has meant thatmany orphan receptor sequences have been finally paired up with atleast one cognate ligand.
Due to the growing evidence that inflammatory cells migrating fromthe circulation to the vascular wall are key players during atherogenes-is, there are increasing reports demonstrating expression and implica-tion of chemokines and their receptors in the atherosclerosis process[52]. Few years ago, the chemokine MCP-1 and IL-8 has been detectedwithin human and animal atherosclerotic lesions, expressed by lesionalmacrophages-derived foam cells and by endothelial cells or SMCs atseveral different stage of the disease [53]. Both of these chemokineswere not present in normal artery. Then, the chemokines MCP-4 andRANTES were also identify within atherosclerotic plaques [54]. In thesame time, the chemokines receptors CCR2 and CXCR1, were alsoidentified at the surface of monocyte-macrophages within lesions. Butthe strong evidence that chemokines and their receptors play importantrole in atherogenesis came in 1998 with two reports using animal model to block the CCR2-MCP-1 pathway. In the first study, the in-vestigators generated a CCR2 knock-out (-/-) mice, which they then crossed to ApoE-/- mice, a well-known and documented mice model of atherosclerosis. Compared to ApoE-/-mice, the CCR2-/- / ApoE-/-present an overall decrease in atherosclerotic lesion size and fewermacrophages were noted in the aortas, without any effect on cholesterolplasma levels [55].
Despite these increasing evidence for involvement of T lymphocytes inatherogenesis, the mechanism of T lymphocyte recruitment within thevascular atherosclerotic lesions remains incompletely defined. In thisprospective, we studied the expression of the three IFNγ-inducible CXCchemokines IP-10, Mig and I-TAC, known to exert potent chemotaxis onactivated-Th1 lymphocyte in other inflammatory dise-ases such as psori-asis or sarcoidosis [56]. All three chemokines signal through a commonreceptor, CXCR3, expressed by activated-Th1 lymphocyte, but not bymonocytes or neutrophils. Using immunohistochem-istry and Western
blot techniques we were able to demonstrate a differential expression ofIP-10, Mig and I-TAC by atheroma associated cells [57]. Interestingly,we found a high expression of the receptor CXCR3 by all T lymphocyteswithin human atherosclerotic lesions. In vitro experiments showed thatthe pro-inflammatory cytokines TNFα and IL-1β, as well as CD40ligand potentate IP-10 expression from IFNγ-stimulated endothelialcells. Moreover, nitric oxide (NO) treatment decreased IFNγ inductionof IP-10. These findings suggest that the differential expression of IP-10,Mig and I-TAC by atheroma associated cells plays a role in the recruit-ment and retention of activated T lymphocytes observed within vascularwall lesion during atherogenesis. We also showed that blocking chemok-ine signalling in vivo through deletion of the chemokine receptors CCR2and CXCR3 has differential effects during atherogenesis with particularconsequences for the regulatory T lymphocytes during early athero-genesis [58]. On the basis of these evidences, we have also investigatedthe role of other chemokines on atherosclerotic plaque development,showing a crucial role for CCR5 (Fig. 3) [59]. Furthermore, we were able to demonstrate for the first time that chronic treatment witha chemokine receptor antagonist (Met-RANTES) limits the progressionof atherosclerosis in vivo [60].
Recent clinical trials showed that C-reactive protein (CRP) is a power-ful independent predictor of future cardiovascular events [61]. CRP isalso the best physiological marker for measuring inflammation, theoccult killer in cardiovascular disease. CRP, the major acute-phasereactant in humans, is mainly produced by hepatocytes in response tointerleukin-6 (IL-6) and is then secreted into systemic circulation. Recent studies reported that, besides its predictive role in determiningcardiovascular risk, CRP could exert a direct pro-atherogenic role. Indeed, it has been shown that CRP accelerates the progression of athe-rosclerosis in Apolipoprotein E-deficient mice [64]. Evidence for thepro-atherogenic role of CRP is further provided by in vitro studiesreporting that CRP modulates the activity and expression of multiplefactors implicated in atherogenesis. CRP downregulates endothelial
nitric oxide synthase (eNOS), resulting in decreased release of NO, and thus facilitation of endothelial cells apoptosis and inhibition of angiogenesis. In addition, CRP stimulates the production of the vasoconstrictor endothelin-1 (ET-1) and the inflammatory marker IL-6 by endothelial cells [63]. Furthermore, CRP increases the ex-pression of vascular cell adhesion molecule-1 (VCAM-1), intercellularadhesion molecule-1 (ICAM-1), E-selectin and monocyte chemo-attractant protein-1 (MCP-1), resulting in enhanced leukocyte trans-migration [64]. The statin connectionIn primary and secondary prevention, the lipid-lowering drug statins(HMG-CoA reductase inhibitor) have shown great efficacy in reducingcardiovascular events and deaths. Since the results of the ScandinavianSimvastatin Survival Study (4S) were first reported over 10 years ago,many other trials have corroborated the beneficial effects of this classof drugs [65]. Some results of these trials indicate that statins may haveanti-inflammatory effects. In 2005, the Pravastatin or AtorvastatinEvaluation and Infection Therapy (PROVE-IT) trial clearly demonstrat-ed that CRP is a marker of CV risk in primary and secondary preven-tion [66]. The PROVE-IT investigators also showed that some statinsmight have greater power to reduce CRP than others. In this trial, ator-vastatin, 80 mg/day, reduced CRP significantly more than pravastatin,40 mg/day, within the remarkably short interval of 30 days. Pleiotropic effects of statins target CRPThere is no doubt that statins have lipid-lowering effects, but theseagents also appear to have pleiotropic effects (beyond their lipid lower-ing properties). We recently demonstrated for the first time that statinsregulate IL-6-induced CRP expression in human hepatocytes, its site ofproduction (Figure 1) [67]. This reduction in CRP expression occursboth at the protein and the mRNA level, indicating that statins exerttheir effect at the transcriptional level. In our in vitro model, statinswere used at doses corresponding to those measured in human plasma(Figure 1). We observed that, similar to the results in humans, ator-vastatin seems to be the most potent inhibitor of CRP release. Althoughnumerous clinical studies have reported that statins lower plasma levels
of CRP, it is the first study showing a direct effect of statins on IL-6-induced CRP expression in hepatocytes. In our study, we also show thatthe statin-mediated reduction of CRP release can be mimicked byGGTI (geranylgeranyl transferase inhibitors). Thus, it seems that theeffect of statins on CRP release occurs via the inhibition of proteingeranylgeranylation. Figure 1Statins reduce IL-6-induced CRP in hepatocytes. CRP release measured by ELISA in supernatants of hepatocytes cultured for 24 hours in nor-mal media (Ctrl=control); activated by 50 ng/mL IL-6 alone (V=vehicle of statin); or in thepresence of pravastatin (Prv 5, 10, and 20 µmol/L); simvastatin (Smv 0.01, 0.1, and 1 µmol/L);atorvastatin (Atv 0.01, 0.1, and 1 µmol/L), or 0.1 µmol/L atorvastatin; and 400 µmol/L L-mevalonate (Meva). *P<0.05 vs IL-6; **P<0.001 vs IL-6; ***P<0.05 vs IL-6+Atv. [67].
In our investigations we also showed that statin pretreatment signi-ficantly reduced STAT3 phosphorylation on serine, but not on tyrosineresidues. The signaling pathways leading to STAT3 phosphorylationare well known and differ among tyrosine or serine residues. In the firstcase, the tyrosine phosphorylation is mediated by the tyrosine kinaseactivity of the stimulated IL-6 receptor complex (IL-6Rα/gp130/JAK). In the other case, recent reports indicate that the serine phosphorylationis mediated by protein kinase C-delta, which in turn is activated by asignal transduction pathway consisting of Vav, Rac-1, MEKK and SEK-1. In our study, we have shown that statins reduce IL-6-inducedCRP expression via the inhibition of protein geranylgeranylation. STAT3 is known to be regulated by Rac-1, a member of the Rhofamily, which needs to be geranylgeranylated to activate downstream
cascade. In conclusion, we hypothesize that, by suppressing the geranyl-geranylation of Rac-1 in hepatocytes, statins reduce IL-6-inducedphosphorylation of STAT3, thus resulting in reduced CRP expression(Figure 2). Such direct anti-inflammatory consequences may improvethe understanding of the clinical effects of statins on cardio-vascularevents and mortality. Figure 2Proposed signaling pathways leading to statin-induced reduction of CRP release in hepa-tocytes (JAK indicates Janus kinase; APRE: acute phase response element; P-STAT3:phospho-STAT3) [67].The endocannabinoid systemThe discovery of membrane receptors that bind the psychoactive comp-ound of marijuana, Δ9-tetrahydrocannabinol (THC) and their endogen-ous ligands has led to the description of the endocannabinoid system. At present, the system is composed of two receptors that have beencloned, and endogenous ligands or endocannabinoids including anand-amide, 2-arachidonoylglycerol, and others. All endocannabinoids ident-ified so far are derivatives of long-chain polyunsaturated fatty acids andexhibit varying selectivity for the two cannabinoid receptors [68].
Both cannabinoid receptors are G protein-coupled receptors that modul-ate second messengers and signaling components such as adenylate
cyclase, mitogen-activated protein kinases or members of the NF-κBfamily. The tissue distribution of the two receptors is likely to accountfor the well-known psychotropic and peripheral effects of THC. Cannabinoid receptor 1 (CB1) is expressed predominantly in the centraland peripheral nervous system, while cannabinoid receptor 2 (CB2) ispresent on immune cells [69]. Thus, CB2 receptors may have physio-logical importance in immune response, inflammation and chronic pain. Immunosuppressive and anti-inflammatory propertiesThe development of selective agonists, antagonists, and transgenicmice lacking CB1 and CB2 receptors has contributed to broaden our cur-rent understanding of cannabinoid biology. As a consequence, the capa-city of cannabinoids to regulate immune function is now well establis-hed. In vitro, THC treatment of human immune cells inhibits secretionof pro-inflammatory cytokines and chemokines and triggers the diffe-rentiation into a Th2 phenotype [70]. As demonstrated, a CB2-specificantagonist abrogates the majority of these immunomodulatory effects[70]. Moreover, THC-mediated inhibition of T helper cell activation isabsent in CB2-deficient mice, supporting the hypothesis that the immu-nomodulatory effects of cannabinoids are CB2-dependent [71].
Cannabidiol, the major non-psychotropic constituent of the Cannabissativa plant, has been reported to ameliorate chronic inflammation inmurine collagen-induced arthritis, a mouse model of rheumatoid arthri-tis, by inhibiting antigen-specific lymphocyte proliferation and interfe-ron-(IFN)-γ secretion [72]. More recently, marijuana-derived cannabi-diol was shown to inhibit macrophage chemotaxis in vitro and in vivoin a CB2 receptor-dependent manner [73]. CB2 is expressed within plaquesTo investigate whether CB2 receptor was expressed in atheroscleroticlesions, we visualized the receptors in human coronary and mouse aortic plaques using immunostaining techniques. We observed abundantCB2 receptor expression in both mouse and human plaques, while nonewas found in normal arteries [74]. Double immunofluorescence stain-ing revealed that the CB2 receptor is mainly expressed on macrophagesand CD4+ T cell lymphocytes within atherosclerotic plaques. Cannabinoids and atherogenesisTo investigate whether THC treatment might reduce the burden ofatherosclerotic disease, we placed apolipoprotein E (apoE)-knockoutmice on a high-fat cholesterol diet. After 5 weeks on the diet, THC wasadministered to the treatment-randomized mice and these were compar-ed to controls 6 weeks later. In the control mice, lesions increased on thehigh-cholesterol diet; however, lesion progression decreased signific-antly in the mice treated with a very low dose of oral THC (Figure 3).
Figure 3Treatment with THC reduces atherosclerotic plaque development in ApoE-/-mice. Atherosclerotic lesions within aortic roots were analysed by Sudan IV staining for lipiddeposition. Quantification of lipid deposition (immunostaining) was performed bycomputer image analysis. *P<0.05 compared with ApoE-/- mice at 5 weeks; **P<0.05compared with ApoE-/- mice at 11 weeks without THC (vehicle) [75].
When we added a specific CB2 receptor blocker, the protective effectof THC was completely abolished, indicating that the atheroprotectionwas due exclusively to the CB2 receptor pathway. T cell activation andproliferation as well as IFN-γ production declined in the lymph nodesof THC treated mice, but no effect was found on anti-inflammatorycytokine production. We also demonstrated reduced migration of perit-oneal cavity macrophages from the same THC-treated apoE-knockoutmice, in vivo, with stimulation by IFN-γ or tumor necrosis factor(TNF)-α. This effect was also clearly linked to the CB2 receptor, sincemigration was unaffected in CB2 knockout mice [75]. CCR2 (ß-sub-
class) receptors are among the most important chemokine receptorsresponsible for macrophage attraction from the circulation to vasculartissue and plaques. CCR2 expression was strongly reduced in splenoc-ytes from apoE-knockout mice treated with THC, in vitro, at dosescorresponding to effective in vivo dosages (Figure 4). This couldexplain the reduction, in vivo, of macrophage migration and atherogen-esis. It is particularly noteworthy that the observed in vitro and in vivoeffects of THC were dose-dependent. The dose dependency showed aU-shaped curve, where both higher and lower doses were inactive. Theeffective dose was lower than the dose usually associated with psycho-tropic effects of THC. Figure 4Treatment with THC reduces CCR2 mRN A expression in vitro. Isolated splenocytes obtained from ApoE-/- mice were stimulated with 10 ng/ml TNF-αin the presence or absence of different doses of THC. Relative expression levels of CCR2messenger RNA were determined by quantitative real-time PCR. Data represent meanvalues + s.e.m. *P<0.05 compared with control, unstimulated cells; **P<0.05 χομπαρεδ
ωιτη TNΦ-α-stimulated cells [75].Cannabinoids as therapeutic agentsOur study has demonstrated that THC, by acting on CB2 receptors,reduces inflammation and the infiltration of immune cells into ather-osclerotic lesions. Nevertheless, these effects are in conflict with theknown adverse effects associated with marijuana consumption. Indeed,the bioactive constituents of the marijuana plant and their synthetic and
endogenous analogs cause not only neurobehavioral, but also cardio-vascular effects as demonstrated in humans and animal models [75, 76]. In humans, the most consistent cardiovascular effects of marijuanasmoking are peripheral vasodilatation and tachycardia. These effectsmanifest themselves as an increase in cardiac output, increased periph-eral blood flow, and variable changes in blood pressure. In anesthetizedrats and dogs, THC produces an acute rise in blood pressure followedby long-lasting hypotension and bradycardia. Based on these hypoten-sive effects, targeting the endocannabinoid system is likely to offer noveltherapeutic strategies in the treatment of hypertension. In a multicenterstudy performed with 3882 patients, marijuana smoking was identifiedas a rare trigger of acute myocardial infarction [77]. While some of thecardiovascular effects result from activation of CB1 receptors, recentfindings indicate that several cannabinoid effects are not mediated byeither CB1 or CB2 receptors [78].
Besides the risk of unwanted cardiovascular effects, a broad acceptanceof cannabinoids as therapeutic agents is hampered by the fact that theyexhibit psychotropic effects. Therefore, particular research interest isfocusing on the development and characterization of either synthetic orplant derived cannabinoids with therapeutic value that are non-psycho-tropic. Among the non-psychotropic plant derived drugs, cannabidiol, amajor compound of marijuana, has been shown to have potent anti-inflammatory and neuroprotective properties, as discussed above [70]. First clinical trials performed with THC/cannabidiol plant extracts havedemonstrated that the study medications were generally well toleratedand associated with only few side effects. However, larger scale andlong-term studies are warranted to confirm a clinically relevant im-provement of neurological symptoms [79].
More interestingly, several non-psychotropic synthetic cannabinoidswith anti-inflammatory properties have recently been developed fromplant cannabinoids, such as ajulemic acid (CT3) and HU-320. Of parti-cular interest is the development of agonists that selectively target CB2receptors. In preclinical studies, CB2 receptor agonists, such as HU308and AM-1241, have been shown to be devoid of effects on the centralnervous system and show promise for the treatment of acute and
chronic pain [80]. A promising novel compound designated Sch.336,which belongs to a new class of CB2-specific ligands, the triaryl bis-sulfones, blocks leukocyte recruitment in vivo [81]. This effect wasdemonstrated in three different models of inflammation. Their anti-inflammatory properties suggest that CB2 ligands may serve as novelimmunomodulatory agents in the treatment of immune disorders such asatherosclerosis. However, little is known about the molecular mode ofaction of these compounds and requires further investigation. Finally, itremains unclear whether receptor signaling via endocannabinoids playsa modulatory role in chronic inflammation ongoing during atherogenesis. A recent report demonstrates that CB1 receptors mediate intrinsic pro-tective signals that counteract pro-inflammatory responses in a mousemodel of colonic inflammation [82]. Additional studies using selectiveCB1 and CB2 receptor antagonists are warranted to investigate a possiblerole of the endocannabinoid system during atherosclerosis.
Inflammatory processes play a pivotal role in all stages of atheroscler-osis. Many risk factors contribute as triggers of inflammatory reactionsand injury to the endothelium. Growing evidence suggests that elevatedplasma levels of vascular wall inflammation markers may help to predictfuture risk of plaque rupture. Prevention and current treatments foratherosclerosis are mainly based on drugs that lower plasma cholesterolconcentration and high blood pressure. In particular, statins have provento reduce the risk of cardiovascular events significantly, not only bytheir cholesterol-lowering properties, but also by their more recentlyidentified anti-inflammatory and immunomodulatory effects. Never-theless, atherosclerosis remains the primary cause for heart disease andstroke. Thus, a great challenge for future research would be the identi-fication and development of promising novel anti-inflammatory thera-pies, such as anti-chemokines or non-psychotropic cannabinoids, toreduce the progression of atherosclerosis. A major challenge of con-temporary medicine is to break the traditional compartmentalizationthat frequently separates different fields. The same holds betweenmedical practice and basic biochemical mechanism. Unexpected link-
ages between different areas of medicine are indeed of particular inter-est. Unsuspected “bridges” across cardiology practice and molecularimmunology, as presented here, are good examples of such a linkage.
I wish to express my deep gratitude to the Prof. Max CloëttaFoundation for the great honour to receive this award.
This prize distinguishes intense research efforts hat could only beachieved by substantial support of highly dedicated technicians, PhDstudents, postdoctoral fellows and other research collaborators frombasic to clinical science. I would like to thank my past and present labteam (especially Fabienne Burger, Sabine Steffens, Graziano Pelli,Vincent Braunersreuther and Fabrizio Montecucco) for their noble end-urance, enthusiasm and contribution to the scientific work honouredhere. I am also very grateful to Professor Peter Libby, Chief ofCardiology at the Brigham and Women’s Hospital, Harvard MedicalSchool Boston, USA, who gave me the opportunity the investigate the vascular biology of atherosclerosis in one of the best scientific en-vironment.
Over the years, I have always been very well supported by several insti-tutions. For their contribution to my research work, I am very gratefulto the Swiss National Science Foundation, the Swiss Heart Foundation,the Novartis Foundation, the Geneva University Faculty of Medicine,the Foundation for Medical Research and the European VascularGenomics Network.
Finally, I would like to express my deep gratitude to my wife Marionand our children Anaëlle, Laurane and Mathieu for their continuingsupport in helping me to maintain the best equilibrium between profes-sional activities and familial relationship, probably the most importantchallenge to many of us.
Lopez A. D., Murray C. C. The global burden of disease, 1990–2020. Nat Med 1998;4:1241–3.
Murray C. J., Lopez A. D. The incremental effect of age-weighting on YLLs, YLDs,and DALYs: a response. Bull World Health Organ 1996; 74:445–6.
Morrow R. H, Hyder A. A, Murray C. J., Lopez A. D. Measuring the burden ofdisease. Lancet 1998; 352:1859–61.
Libby P. Inflammation in atherosclerosis. Nature 2002; 420:868–74.
Virchow R. Der Ateromatose Prozess der Arterien. Wien Med Wochenschr 1856;6:825–841.
Hansson G. K., Libby P. The immune response in atherosclerosis: a double-edgedsword. Nat Rev Immunol 2006; 6:508–519.
Breslow J. L. Mouse models of atherosclerosis. Science 1996; 272:685–8.
Sullivan G. W, Sarembock I. J., Linden J. The role of inflammation in vasculardiseases. J Leukoc Biol 2000; 67:591–602.
Springer TA CM. Traffic signals on endothelium for leukocytes in health, inflamma-tion, and atherosclerosis. In: Fuster V., Ross R., Topol E. J., eds. Atherosclerosis andcoronary artery disease. 1996; 1:595–606.
10. Picker L. J., Butcher E. C. Physiological and molecular mechanisms of lymphocyte
homing. Annu Rev Immunol 1992; 10:561–91.
11. Picker L. J. Mechanisms of lymphocyte homing. Curr Opin Immunol 1992; 4:277–86.
12. Baggiolini M. Chemokines and leukocyte traffic. Nature 1998; 392:565–8.
13. Ross R. Atherosclerosis: An inflammatory disease. N Engl J Med 1999; 340:115–126.
14. Glass C. K., Witztum J. L. Atherosclerosis. the road ahead. Cell 2001; 104:503–16.
15. Lusis A. J. Atherosclerosis. Nature 2000; 407:233– 41.
16. Libby P. Inflammation in atherosclerosis. Nature 2002; 420:868–74.
17. Libby, P., P. M. Ridker, and A. Maseri, Inflammation and atherosclerosis. Circulation
18. De Caterina R., Libby P., Peng H. B. et al. Nitric oxide decreases cytokine-induced
endothelial activation. Nitric oxide selectively reduces endothelial expression ofadhesion molecules and proinflammatory cytokines. J Clin Invest 1995; 96:60–68.
19. Nagel T., Resnick N., Atkinson W. J., et al. Shear stress selectively upregulates inter-
cellular adhesion molecule-1 expression in cultured human vascular endothelialcells. J Clin Invest 1994; 94:885–91.
20. Gu L., Okada Y., Clinton S., et al. Absence of monocyte chemoattractant protein-1
reduces atherosclerosis in low-density lipoprotein-deficient mice. Mol Cell 1998;2:275–81.
21. Boring L., Gosling J., Cleary M., et al. Decreased lesion formation in CCR2-/- mice
reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998;394:894–7.
22. Veillard N. R., Kwak, Pelli G., Mulhaupt F., James R.W., Proudfoot AEI, Mach F.
Antagonism of RANTES Receptors Reduces Atherosclerotic Plaque Formation inMice Circ Res 2004; 94:253–61.
23. Sheikine Y., Hansson G. K. Chemokines and atherosclerosis. Ann Med 2004; 36:98–118.
24. Smith J. D., Trogan E., Ginsberg M., et al. Decreased atherosclerosis in mice deficient
in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc NatlAcad Sci U S A 1995; 92:8264–68.
25. Hansson G., Libby P. The role of the lymphocyte. In: Fuster V., Ross R., Topol E.,
eds. Atherosclerosis and Coronary Artery Disease. New York, NY: Lippincott-Raven1996; 557–68.
26. Clark L. B., Foy T. M., Noelle R. J. CD40 and its ligand. Adv Immunol 1996; 63:43–78.
27. Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev
28. Van Kooten C., Banchereau J. CD40-CD40 ligand: a multifunctional receptor-ligand pair. Adv Immunol 1996; 61:1–77.
29. Noelle R. J., Ledbetter J. A., Aruffo A. CD40 and its ligand, an essential ligand-recep-
tor pair for thymus- dependent B-cell activation. Immunol Today 1992; 13:431–3.
30. Noelle R. J. CD40 and its ligand in cell-mediated immunity. Agents Actions Suppl
31. Noelle R. J., Mackey M., Foy T., Buhlmann J., Burns C. CD40 and its ligand in auto-
immunity. Ann N Y Acad Sci 1997; 815:384–91.
32. Callard R. E., Armitage R. J., Fanslow W. C., Spriggs M. K. CD40 ligand and its role
in X-linked hyper-IgM syndrome. Immunol Today 1993; 14:559–64.
33. Callard R. E., Smith S. H., Herbert J., et al. CD40 ligand (CD40L) expression and B
cell function in agammaglobulinemia with normal or elevated levels of IgM (HIM). Comparison of X-linked, autosomal recessive, and non-X-linked forms of the disease,and obligate carriers. J Immunol 1994; 153:3295–306.
34. Schonbeck U., Mach F., Libby P. CD154 (CD40 ligand). Int J Biochem Cell Biol
35. Durie F. H., Fava R. A., Noelle R. J. Collagen-induced arthritis as a model of
rheumatoid arthritis. Clin Immunol Immunopathol 1994; 73:11–8.
36. Mohan C., Shi Y., Laman J. D., Datta S. K. Interaction between CD40 and its ligand
gp39 in the development of murine lupus nephritis. J Immunol 1995; 154:1470–80.
37. Gerritse K., Laman J. D, Noelle R. J., et al. CD40-CD40 ligand interactions in ex-
perimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad SciUSA 1996; 93:2499–504.
38. Larsen C. P., Elwood E. T, Alexander D. Z., et al. Long-term acceptance of skin and
cardiac allografts after blocking CD40 and CD28 pathways. Nature 1996; 381:434–8.
39. Larsen C. P., Alexander D. Z., Hollenbaugh D., et al. CD40-gp39 interactions play a
critical role during allograft rejection. Suppression of allograft rejection by blockadeof the CD40-gp39 pathway. Transplantation 1996; 61:4–9.
40. Durie F. H., Aruffo A., Ledbetter J., et al. Antibody to the ligand of CD40, gp39,
blocks the occurrence of the acute and chronic forms of graft-vs-host disease. J ClinInvest 1994; 94:1333–8.
41. Mach F., Schonbeck U., Sukhova G. K., Bourcier T., Bonnefoy J. Y., Pober J. S.,
Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells,smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signal-ing in atherosclerosis. Proc Natl Acad Sci U S A 1997; 94:1931–1936.
42. Mach F., Schonbeck U., Bonnefoy J. Y., Pober J. S., Libby P. Activation of monocyte/
macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation 1997;96:396–399.
43. Schonbeck U., Mach F., Bonnefoy J. Y., Loppnow H., Flad H. D., Libby P. Ligation
of CD40 activates interleukin 1beta-converting enzyme (caspase-1) activity invascular smooth muscle and endothelial cells and promotes elaboration of activeinterleukin 1beta. J Biol Chem. 1997; 272:19569–19574.
44. Schonbeck U., Mach F., Sukhova G. K., Murphy C., Bonnefoy JY, Fabumni RP, Libby
P. Regulation of matrix metalloproteinase expression in human vascular smoothmuscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? CircRes. 1997; 81:448–454.
45. Mach F., Schonbeck U., Sukhova G. K., Atkinson E., Libby P. Reduction of athero-
sclerosis in mice by inhibition of CD40 signalling. Nature 1998; 394:200–203.
46. Baggiolini M., Dewald B., Moser B. Human chemokines: an update. Annu Rev
47. Baggiolini M., Loetscher P. Chemokines in inflammation and immunity. Immunol
48. Schall T. J., Bacon K. B. Chemokines, leukocyte trafficking, and inflammation. CurrOpin Immunol 1994; 6:865–73.
49. Schall T. J., Mak J. Y., DiGregorio D., Neote K. Receptor/ligand interactions in the
C-C chemokine family. Adv Exp Med Biol 1993; 351:29–37.
50. Baggiolini M., Dewald B., Moser B. Interleukin-8 and related chemotactic cytok-
ines--CXC and CC chemokines. Adv Immunol 1994; 55:97–179.
51. Baggiolini M., Dahinden C. A. CC chemokines in allergic inflammation. Immunol
52. Gerszten R. E., Mach F., Sauty A., Rosenzweig A., Luster A. D. Chemokines, leuk-
ocytes, and atherosclerosis. J Lab Clin Med 2000; 136:87–92.
53. Boisvert W. A., Curtiss L. K., Terkeltaub R. A. Interleukin-8 and its receptor CXCR2
in atherosclerosis. Immunol Res 2000; 21:129–37.
54. Berkhout T. A., Sarau H. M., Moores K., et al. Cloning, in vitro expression, and
functional characterization of a novel human CC chemokine of the monocytechemotactic protein (MCP) family (MCP-4) that binds and signals through the CCchemokine receptor 2B. J Biol Chem 1997; 272:16404–13.
55. Boring L., Gosling J., Cleary M., Charo I. F. Decreased lesion formation in CCR2-/-
mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998;394:894–7.
56. Gottlieb A. B., Luster A. D., Posnett D. N., Carter D. M. Detection of a gamma
interferon-induced protein IP-10 in psoriatic plaques. J Exp Med 1988; 168:941–8.
57. Mach F., Sauty A., Sukhova G. K., Libby P., Luster A. D. The interferon-gamma
inducible CXC-chemokines IP-10, Mig and I-TAC are expressed by human atheroma-associated cells: Implication for lymphocyte recruitment in atherogenesis. J. Clin. Invest. 1999; 104:1041–1050.
58. Veillard N., Steffens S., Pelli G., Lu B., Kwak B. R, Gerard C., Charo I. F, Mach F.
Differential influence of chemokine receptors CCR2 and CXCR3 in development ofatherosclerosis in vivo. Circulation 2005; 112:870–878.
59. Braunersreuther V., Zernecke A., Arnaud C., Liehn E. A., Steffens S, Shargdarsuren
E., Bidzheckov K., Burger F., Pelli G., Luckow V., Mach F., Weber C. Ccr5 but notCcr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2007; 27:373-379.
60. Veillard N, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AEI, Mach F.
Antagonist of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004; 94:253–261.
61. Ridker P. M. Clinical application of C-reactive protein for cardiovascular disease
detection and prevention. Circulation 2003; 107:363–369.
62. Paul A., Ko K. W., Li L., Yechoor V., McCrory M. A., Szalai A. J., Chan L. C-reacti-
ve protein accelerates the progression of atherosclerosis in apolipoprotein E-deficientmice. Circulation 2004; 109:647–655.
63. Verma S., Li S. H., Badiwala M. V, Weisel R. D., Fedak P. W, Li R. K., Dhillon B.,
Mickle D. A. Endothelin antagonism and interleukin-6 inhibition attenuate theproatherogenic effects of C-reactive protein. Circulation 2002; 105:1890–1896.
64. Pasceri V., Cheng J. S., Willerson J. T., Yeh E. T. Modulation of C-reactive protein-
mediated monocyte chemoattractant protein-1 induction in human endothelial cellsby anti-atherosclerosis drugs. Circulation 2001; 103:2531–2534.
65. Scandinavian Simvastatin Survival Study (4S). Group.Randomised trial of cholest-
erol lowering in 4444 patients with coronary heart disease: the ScandinavianSimvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
66. Ridker P. M, Cannon C. P, Morrow D., Rifai N., Rose L. M., McCabe C. H., Pfeffer
M. A., Braunwald E; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28.
67. Arnaud C., Burger F., Steffens S., Veillard N. R., Nguyen T. H., Trono D., Mach F.
Statins reduce interleukin-6-induced C-Reactive Protein in human hepatocytes. Newevidence for direct anti-inflammatory effects of statins. Arterioscler. Thromb. Vasc. Biol. 2005; 25:1231–1236.
68. McAllister S. D., Glass M. CB(1) and CB(2) receptor-mediated signalling: a focus on
endocannabinoids. Prostaglandins Leukot Essent Fatty Acids 2002; 66:161–171.
69. Lutz B. Molecular biology of cannabinoid receptors. Prostaglandins Leukot EssentFatty Acids 2002; 66:123–142.
70. Yuan M., Kiertscher S. M., Cheng Q., Zoumalan R., Tashkin D. P., Roth M. D. Delta
9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human Tcells. J Neuroimmunol. 2002; 133:124–131.
71. Buckley N. E., McCoy K. L., Mezey E., Bonner T., Zimmer A., Felder C. C., Glass
M., Zimmer A. Immunomodulation by cannabinoids is absent in mice deficient forthe cannabinoid CB(2) receptor. Eur J Pharmacol. 2000; 396:141–149.
72. Malfait A. M., Gallily R., Sumariwalla P. F., Malik A. S., Andreakos E, Mechoulam
R., Feldmann M. The nonpsychoactive cannabis constituent cannabidiol is an oralanti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci US A. 2000; 979561–979566.
73. Sacerdote P., Martucci C., Vaccani A., Bariselli F., Panerai A. E., Colombo A.,
Parolaro D., Massi P. The nonpsychoactive component of marijuana cannabidiolmodulates chemotaxis and IL-10 and IL-12 production of murine macrophages bothin vivo and in vitro. J Neuroimmunol. 2005; 159:97–105.
74. Steffens S., Veillard N. R., Arnaud C., Pelli G., Burger F., Staub C., Karsak M.,
Zimmer A., Frossard J. L., Mach F. Low dose oral cannabinoid therapy reducesprogression of atherosclerosis in mice. Nature. 2005; 435:528.
75. Randall M. D., Kendall D. A., O’Sullivan S. The complexities of the cardiovascular
actions of cannabinoids. Br J Pharmacol. 2004; 142:20–26.
76. Pacher P., Batkai .S, Kunos G. Blood pressure regulation by endocannabinoids and
their receptors. Neuropharmacology 2005; 48:1130–1138.
77. Mittleman M. A., Lewis R. A., Maclure M., Sherwood J. B., Muller J. E. Triggering
myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
78. Begg M., Pacher P., Batkai S., Osei-Hyiaman D., Offertaler L., Mo F. M., Liu J.,
Kunos G. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145.
79. Berman J. S., Symonds C., Birch R. Efficacy of two cannabis based medicinal
extracts for relief of central neuropathic pain from brachial plexus avulsion: resultsof a randomised controlled trial. Pain 2004; 112:299–306.
80. Ibrahim M. M., Porreca F., Lai J., Albrecht P. J., Rice FL, Khodorova A., Davar G.,
Makriyannis A., Vanderah T. W., Mata H. P., Malan T. P. Jr. CB2 cannabinoid recep-tor activation produces antinociception by stimulating peripheral release of endoge-nous opioids. Proc Natl Acad Sci U S A. 2005; 102:3093–3098.
81. Lunn C. A, Fine J. S., Rojas-Triana A., Jackson J. V., Fan X., Kung T. T., Gonsiorek
W., Schwarz M. A., Lavey B., Kozlowski J. A., Narula S. K., Lundell D. J., HipkinR. W., Bober L. A. A novel cannabinoid CB2 receptor-selective inverse agonistblocks leukocyte recruitment in vivo. J Pharmacol Exp Ther. 2006; 31:780–788.
82. Massa F., Marsicano G., Hermann H., Cannich A., Monory K., Cravatt B. F., Ferri
G. L., Sibaev A., Storr M., Lutz B. The endogenous cannabinoid system protectsagainst colonic inflammation. J Clin Invest. 2004; 113:1202–1209.
Skin Blends FAT Burning Drops are to be used in conjunction with the Low Calorie Diet Protocol. You can expect weight loss of 1/2 - 1 lb per day by eating a diet of a variety of lean protein, vegetables and fruit while using your weight loss drops For best results place 10 drops under your tongue (sublingual) about 15 minutes before you eat a meal, 3 times a day. Skin Blends FAT Burn
Top 100 Most Cited Publications in Psychiatry&Psychology_Prepared by Alex J Mitchell using Web of Science (accurate as of 20th Jan 2011) 1st AUTHOR CITES / YR MOVEMENT Mini-mental state - practical method for grading cognitive state of patients for clinician The moderator mediator variable distinction in social psychological-research - conceptual, strategic, and statistical considera
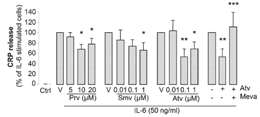 of CRP, it is the first study showing a direct effect of statins on IL-6-induced CRP expression in hepatocytes. In our study, we also show thatthe statin-mediated reduction of CRP release can be mimicked byGGTI (geranylgeranyl transferase inhibitors). Thus, it seems that theeffect of statins on CRP release occurs via the inhibition of proteingeranylgeranylation.
of CRP, it is the first study showing a direct effect of statins on IL-6-induced CRP expression in hepatocytes. In our study, we also show thatthe statin-mediated reduction of CRP release can be mimicked byGGTI (geranylgeranyl transferase inhibitors). Thus, it seems that theeffect of statins on CRP release occurs via the inhibition of proteingeranylgeranylation.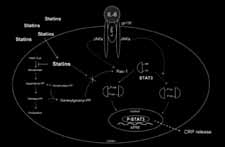 cascade. In conclusion, we hypothesize that, by suppressing the geranyl-geranylation of Rac-1 in hepatocytes, statins reduce IL-6-inducedphosphorylation of STAT3, thus resulting in reduced CRP expression(Figure 2). Such direct anti-inflammatory consequences may improvethe understanding of the clinical effects of statins on cardio-vascularevents and mortality.
cascade. In conclusion, we hypothesize that, by suppressing the geranyl-geranylation of Rac-1 in hepatocytes, statins reduce IL-6-inducedphosphorylation of STAT3, thus resulting in reduced CRP expression(Figure 2). Such direct anti-inflammatory consequences may improvethe understanding of the clinical effects of statins on cardio-vascularevents and mortality.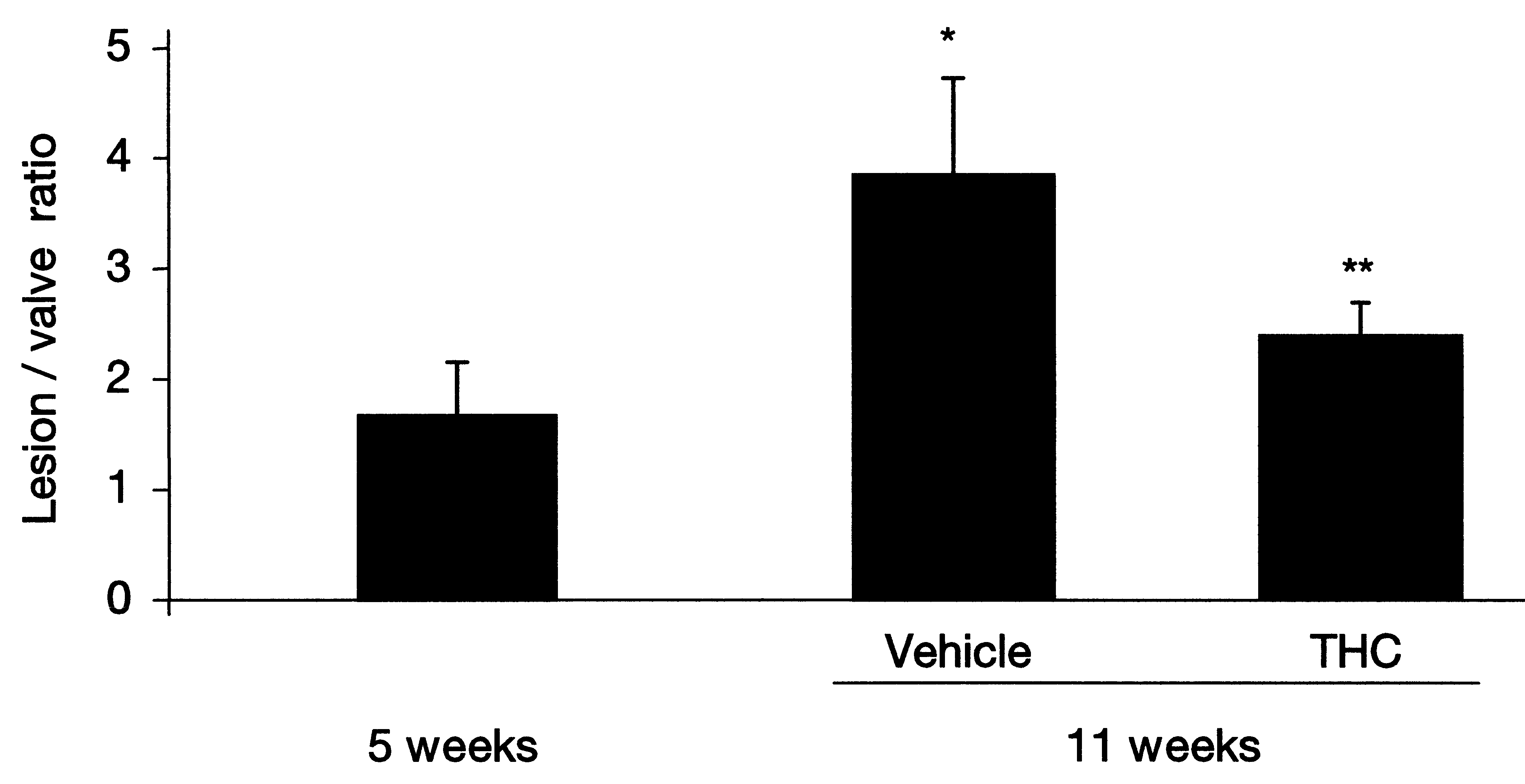
 Cannabinoids and atherogenesisTo investigate whether THC treatment might reduce the burden ofatherosclerotic disease, we placed apolipoprotein E (apoE)-knockoutmice on a high-fat cholesterol diet. After 5 weeks on the diet, THC wasadministered to the treatment-randomized mice and these were compar-ed to controls 6 weeks later. In the control mice, lesions increased on thehigh-cholesterol diet; however, lesion progression decreased signific-antly in the mice treated with a very low dose of oral THC (Figure 3).
Figure 3Treatment with THC reduces atherosclerotic plaque development in ApoE-/-mice.
Cannabinoids and atherogenesisTo investigate whether THC treatment might reduce the burden ofatherosclerotic disease, we placed apolipoprotein E (apoE)-knockoutmice on a high-fat cholesterol diet. After 5 weeks on the diet, THC wasadministered to the treatment-randomized mice and these were compar-ed to controls 6 weeks later. In the control mice, lesions increased on thehigh-cholesterol diet; however, lesion progression decreased signific-antly in the mice treated with a very low dose of oral THC (Figure 3).
Figure 3Treatment with THC reduces atherosclerotic plaque development in ApoE-/-mice.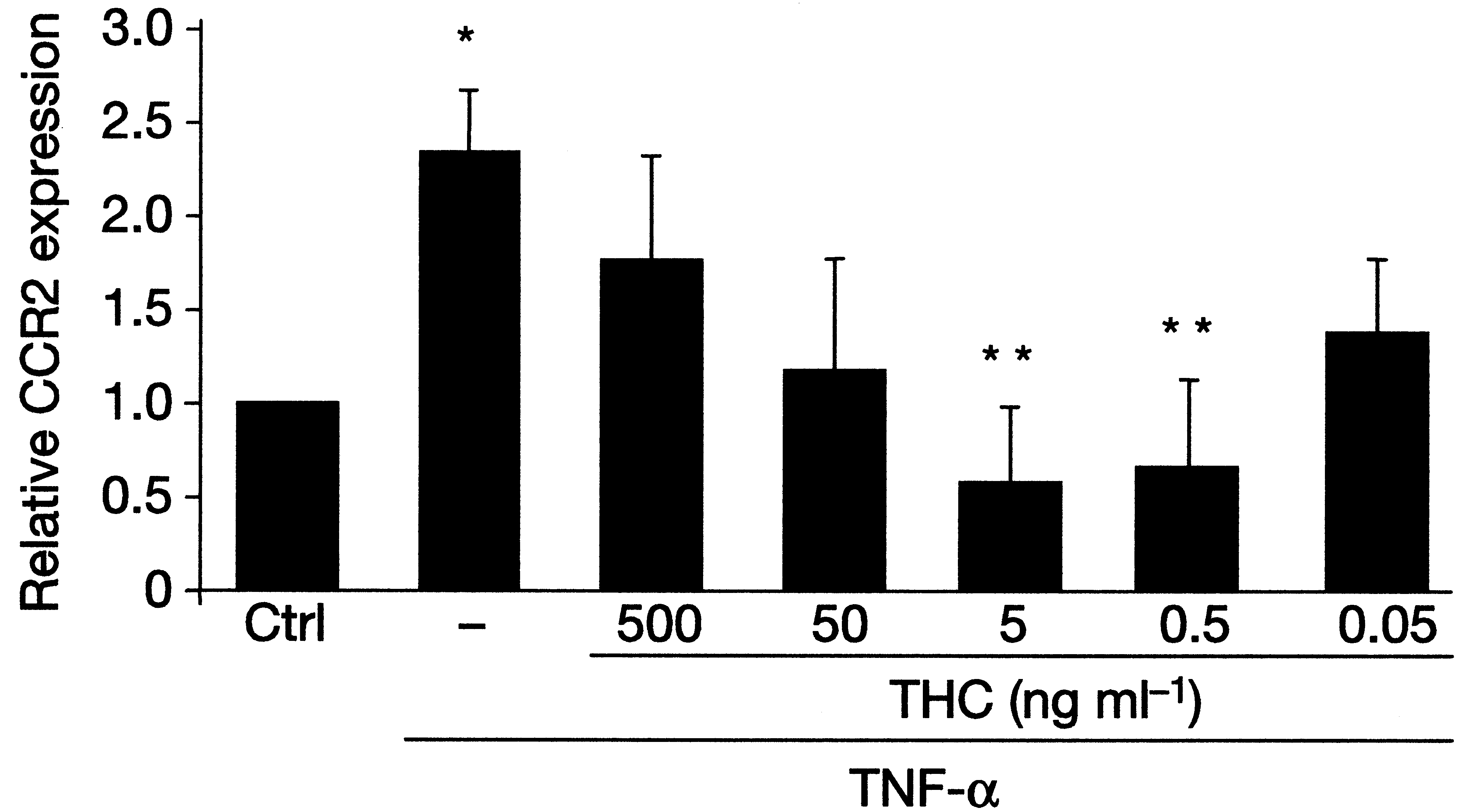 class) receptors are among the most important chemokine receptorsresponsible for macrophage attraction from the circulation to vasculartissue and plaques. CCR2 expression was strongly reduced in splenoc-ytes from apoE-knockout mice treated with THC, in vitro, at dosescorresponding to effective in vivo dosages (Figure 4). This couldexplain the reduction, in vivo, of macrophage migration and atherogen-esis. It is particularly noteworthy that the observed in vitro and in vivoeffects of THC were dose-dependent. The dose dependency showed aU-shaped curve, where both higher and lower doses were inactive. Theeffective dose was lower than the dose usually associated with psycho-tropic effects of THC.
class) receptors are among the most important chemokine receptorsresponsible for macrophage attraction from the circulation to vasculartissue and plaques. CCR2 expression was strongly reduced in splenoc-ytes from apoE-knockout mice treated with THC, in vitro, at dosescorresponding to effective in vivo dosages (Figure 4). This couldexplain the reduction, in vivo, of macrophage migration and atherogen-esis. It is particularly noteworthy that the observed in vitro and in vivoeffects of THC were dose-dependent. The dose dependency showed aU-shaped curve, where both higher and lower doses were inactive. Theeffective dose was lower than the dose usually associated with psycho-tropic effects of THC.