Kamagra enthält Sildenafilcitrat als pharmakologisch aktiven Bestandteil. Dieser hemmt selektiv die Phosphodiesterase-5 und erhöht dadurch die Konzentration von cGMP im Corpus cavernosum. Der Effekt ist zeitlich begrenzt, da die Halbwertszeit von Sildenafil etwa vier Stunden beträgt. In der galenischen Form als Mundgel erfolgt die Resorption besonders rasch, was zu einem schnelleren Wirkeintritt führt. Der Abbau erfolgt überwiegend hepatisch über CYP3A4, wobei ein aktiver Metabolit entsteht, der zur Gesamtwirkung beiträgt. Typische Nebenwirkungen ergeben sich aus der Vasodilatation, darunter leichte Kopfschmerzen und nasale Kongestion. In klinischen Beschreibungen wird kamagra oral jelly im Zusammenhang mit der schnelleren Absorption erwähnt.
Microsoft word - be sublingual protocol.doc
Particulars of the project and investigator(s)
Project No.:A65037-2 EUDRACT No.: 2008-003860-21
1. Project A comparative pharmacokinetic study of sublingual misoprostol after oral mifepristone in pregnant women. 2. Principal investigator(s)
Name(s) : Dr. Oskari Heikinheimo Position : Associate Professor, Senior Physician Postal address : Department of Obstetrics and Gynaecology, Helsinki University Central Hospital PO Box 140, 00029-HUS, FINLAND Telephone : +358 40 5871070
3. Institution responsible for the research project
Name: Department of Obstetrics and Gynaecology, Postal address: Helsinki University Central Hospital PO Box 140, 00029-HUS, FINLAND Telephone: +358 40 5871070 Fax: +358 9 471 74801 E-mail: oskari.heikinheimo@helsinki.fi
5. Responsible financial
Name: Seppo Pakkala Address: Clinical Research Institute HUCH Ltd., Helsinki, Finland Name of institute’s bank: Nordea Bank Bank address: Helsinki, Finland Bank account number:
6. Duration of project
Earliest starting date (if applicable): 1 February 2009 Total (years): One
7. Funds requested (€)
8. Approval of ethics committees
institutional ethical clearance document attached?
B. Is documentation of Finnish Drug Regulatory Agency approval attached?
9. Acceptance responsibility
If this application is accepted, I (we) declare that I (we) shall be actively engaged in, and shall be in day-
to-day control of, the project; and I (we) agree to provide detailed annual progress reports to WHO of the work undertaken. I (we) agree to follow the policies of the Special Programme on the dissemination of research results. The research conducted will conform to the international guidelines for biomedical research involving human subjects and the international principles for biomedical research involving animals.
Signature(s) of principal investigator(s) and date:
_____________________________________________
__Oskari Heikinheimo___________________________
10. Declaration(s) of the Director of the institution (or a designated representative) and the officer responsible for the administration of funds awarded (e.g. Finance Officer, Bursar, etc.)
We confirm that this application has been approved by our institution and that, if granted, the work will be administered in the institution in accordance with its general conditions. The staff gradings and salaries provided are correct and in accordance with the normal practice of the institution. Head of the institution Administrative authority
Name and initials Name and initials (please print) Maija Haukkamaa (please print) Seppo Pakkala_______________
Title Administrative Head_____________ Title Director____________________________
Department of Obstetrics and Gynecology__ Clinical Research Institute HUCH Ltd_________
Date _________________________ Date ________________________________
Signature __________________________ Signature_______________________________
Project Summary Justification for the project
The present study aims at studying the pharmacokinetics of misoprostol following sublingual administration. The original formulation of misoprostol will be compared with a new generic misoprostol formulation that is co-packaged with mifepristone. It is necessary to demonstrate bioequivalence to regulatory authorities.
Proposed research
The proposed project is a pharmacokinetic study that involves a minimum of 60 women with a duration of pregnancy up to nine weeks. Mifepristone (200mg) will be administered orally. Twenty four hours later two doses of 400 μg of misoprostol will be administed at 20 minutes interval sublingually. Blood samples will be collected for 6 hours following the administration of misoprostol. The assay of misoprostol acid will be undertaken and various pharmacokinetic parameters calculated.
features
This will allow the registration of sublingual use of a generic preparation of misoprostol, as the preferred prostaglandin for use with mifepristone for medical abortion.
Technique and skill
The study requires the clinical facilities and expertise to carry out medical abortion, venepuncture, preparation of blood samples and analysis of misoprostol.
Problems anticipated
Form 3. Description of the project Note: Start the description of the project on this page and then continue on additional sheets as necessary. 3.1
3.1.1 Rationale The use of mifepristone together with misoprostol has become the most common method of medical abortion. In the USA and many countries in Europe, the registered dose of mifepristone is the original dose of 600mg used with a prostaglandin. However, given the evidence from multicentred studies (WHO, 2002), in most countries, a significantly lower dose of mifepristone (200mg) is used, followed by 4x200μg of misoprostol given intravaginally. For example, this is the regimen recommended by the Royal College of Obstetricians and Gynaecologists (RCOG) in the United Kingdom (RCOG, 2004). Misoprostol is now the most common prostaglandin to be used in combination with mifepristone and has been shown to be more effective and better tolerated when administered vaginally rather than orally (El-Refaey, 1995; WHO, 2003). Moreover, in a review of outcomes in 4,132 women in Aberdeen, when a dose of 800μg of misoprostol is given intravaginally, 3/1000 had a continuing pregnancy (Ashok, 2002). A similar rate is seen from WHO multicentred studies, while in the same studies, there was a rate of 12/1000 continuing live pregnancies is found after orally administered misoprostol (800μg initial dose, followed by 800μg daily for 7 days) (WHO, 2003). Furthermore, vaginal administration has significantly less side effects. This makes intravaginal administration safer in situations where there is no backup with vacuum aspiration available. Although intravaginal administration of misoprostol is the currently approved and most widely used regimen in early abortion, alternative routes of administration (namely sublingual and buccal) are being researched. Two studies (Tang OS et al, 2003; Hamoda H et al, 2003 & 2005) suggest that sublingual administration of misoprostol may be even more effective than vaginal administration. Moreover, there were no continuing pregnancies in the sublingual groups and complete abortion rates were higher than in the vaginal groups. However, prostaglandin-related side-effects were greater with sublingual use than with vaginal administration of misoprostol. WHO is currently carrying out a randomized placebo-controlled 15-centre trial to compare two doses (400µg and 800µg) and two routes of administration (sublingual and vaginal) of misoprostol after pre-treatment with mifepristone (200mg) for early pregnancy termination (up to 9 completed weeks of gestation). It is anticipated that some 3000 women will be recruited into the study for the study and the final results should be available soon after its completion in July 2008. Being able to offer the choice of sublingual and vaginal administration of misoprostol not only will provide an additional choice to women but also possibility of an even more effective regimen. The World Health Organization has previously licensed intellectual property, in the form of clinical trial data to the non-profit organization, the Concept Foundation, Bangkok. Under this agreement, the Concept Foundation committed to find a high quality pharmaceutical company which will manufacture a copackaged product containing 200mg of mifepristone and 800μg of misoprostol, which can be provided to the public sector of developing countries at a price of less than US$4. Concept Foundation has achieved WHO’s requirements and together with Sun Pharma, the fifth largest pharmaceutical manufacturer in India has been developin a full ICH-compliant registration dossier that for submission to regulatory authorities in both developed and developing countries. As part of this work, a bioequivalence study was undertaken in Helsinki in which 69 women requesting medical termination of an early (≤9 weeks of pregnancy) unwanted pregnancy were randomized to receive either the original mifepristone tablets, Mifegyne, from Exelgyn and the original misoprostol tablets, Cytotec, from Searle or mifepristone and misoprostol, manufactured and co-packaged by Sun Pharma, India. This study has been completed and the pharmacokinetic data are being analyzed. The new study builds on this study and evaluates the sublingual administration of misoprostol with the same two formulations of misoprostol.
3.1.2 Objective To demonstrate compare the pharmacokinetics of misoprostol following sublingual administration
of a new generic misoprostol (co-packaged with mifepristone) with the original formulation of misoprostol.
No comparative pharmacokinetic study comparing sublingual administration of generic and the original formulation of misoprostol, administered following oral mifepristone, for medical abortion has been undertaken. 3.3 Design
A minimum of 60 women will be recruited into the study from those women requesting termination of pregnancy by medical abortion at the Helsinki University Central Hospital. The hospital undertakes some 1,200 medical abortions a year. The objectives of, and procedures involved in, the study will be discussed with them. Mifepristone (200mg) will be administered orally on day 1. At 24 hours, 2 tablets of 200μg tablets of misoprostol will be administered sublingually followed by another 2 tablets of 200 μg 20 minutes later. Blood samples will collected at 15 minute intervals for two hours and then at 3, 4 and 6 hours after misoprostol administration, calculated from the first dose. The pharmacy of the Helsinki University Central Hospital will be responsible for the storage, packaging, randomization and distribution of the study medications. 3.3.2 Inclusion
Subjects admitted to the study will fulfil the following criteria: • good general health
• on day one of the study (day of mifepristone administration) the duration of pregnancy is
not more than 63 days (counted from the first day of the last menstrual period) in a normal 28-day cycle
• the duration of the pregnancy corresponds to the length of amenorrhea when verified by
ultrasound, if the gestational length according to ultrasound measurements differ by more than four days, the ultrasound dating will be used
• the pregnancy is single and intrauterine (single sac)
• if treatment with misoprostol should fail, subject agrees to surgical termination of
• willing and able to participate in the study once the objective and study requirements have
3.3.3 Exclusion criteria: Subjects will not be recruited if any of the following conditions are present:
• allergy towards mifepristone or misoprostol
• a history or evidence of disorders that represent a contraindication to the use of mifepristone (chronic adrenal failure, severe asthma uncontrolled by corticosteroid therapy, inherited porphyria)
• a history or evidence of disorders that represent a contraindication to the use of
prostaglandins (mitral stenosis, glaucoma, sickle cell anaemia, diastolic pressure over 90 mmHg, bronchial asthma, arterial hypotension)
• a history or evidence of thrombo-embolism, severe or recurrent liver disease or pruritus of
• has any medical condition or disease that requires regular treatment with systemic drugs,
care or precaution in conjunction with abortion
• tendency of abnormal bleeding (such as von Willebrandt’s disease)
• previous surgery of uterus/uterine cervix is a relative contraindication, however, previous
low-segment caesarean section is not a contraindication
• suspicion of any pathology of pregnancy (eg, molar, non-viable pregnancy, threatened
• suspected or known breast or genital neoplasia
• where difficulties are anticipated in follow-up.
Women older than 35 years, who smoke less than 20 cigarettes per day, have diastolic pressure of less than 90 mmHg and who have no known risk factor for cardiovascular disease, can be recruited into the study.
In addition, subjects who have been admitted into the study, will be excluded from the final
• the use of drugs, other than those prescribed by the investigator for the treatment of
possible therapy-related side-effects. Pain will be managed with premedication of 1g paracetamol and 30mg codeine, if required, NSAIDs or opiates will be given for additional pain medication
• essential data missing from the subject’s records making it impossible to judge the
• the presence of an acute illness of any nature during the treatment period.
The subjects will be randomized by computer-generated random numbers into two groups. The
random table will be generated by the Head Pharmacist, Helsinki University Central Hospital. Group A will comprise women who will be given 200mg of mifepristone produced by Exelgyn, followed 24 hours later by two doses of 2 tablets of 200μg misoprostol produced by Pfizer. Group B will comprise women who will be given 200mg of mifepristone and two doses of 2 tablets of 200μg misoprostol produced by Sun Pharma.
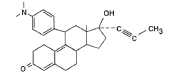
3.3.5 Description of the drugs to be used International Non-proprietary Name: Mifepristone (Mifegyne, Exelgyn and Medabon, Sun Pharma) Chemical name: 11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl) estra-4,9-dien-3-one
Route of administration: oral Amount: 1 x 200mg tablet International Non-proprietary Name: Misoprostol (Cytotec, Pfizer and Medabon, Sun Pharma)
Chemical name: 7-[3-hydroxy-2-(4-hydroxy-4-methyl-oct-1-enyl)-5-oxo-cyclopentyl] heptanoic acid
Route of administration: sublingual Amount: 4 x 200 μg tablets
3.3.6 Admission procedure, treatment and sampling schedule
Before admission to the study, potential subjects will undergo the following:
• ultrasound examination to verify the length of the pregnancy and check that the pregnancy
is intrauterine. If the pregnancy is more advanced, if the there is a suspicion of an abnormal pregnancy (eg, extra-uterine, molar, etc) the woman will not be admitted into the study
• medical, obstetrical and gynaecological history
• medical and gynaecological examination
Women who fulfil the admission criteria, are willing and able to participate in the study and have given their signed informed consent will be included in the study. The study will be performed over a period of two days during the medical termination of pregnancy. Day one Baseline blood pressure, pulse rate, body height and weight will be taken. Medical and obstetrical history, as well as informed consent, will be obtained. The subjects will be randomized by computer-generated random numbers into 2 groups. Each woman will then be given 200mg of mifepristone orally. Following ingestion of mifepristone women are free to leave the hospital. Day two (24h after oral administration of mifepristone) An intravenous catheter suitable for multiple blood sampling will be inserted. A venous blood sample (10ml) will be collected before misoprostol administration; this will be the 0min sample prior to misoprostol. Two tablets of 200μg misoprostol will then be administered sublingually. After 20 minutes the tablets can be swallowed if they have not melted and another two tablets will be administered sublingually. Venous blood samples (10ml) will be taken at 15, 30, 45, 60, 75, 90 and 105min and 2, 3, 4 and 6 hours after administration of misoprostol. All blood samples will be centrifuged without delay and serum will be immediately frozen and stored at -20ºC. Blood pressure, pulse and any reported side effects will be monitored. The interval from the administration of misoprostol to onset of abdominal pain/vaginal bleeding and passage of production of conception will be recorded. The research nurse will stay with the subjects throughout the blood taking procedure to provide emotional support and counselling if necessary. It is anticipated that some 90% of subjects will have aborted by six hours after the administration of misoprostol. Some subjects will abort before six hours, in which case no further blood samples will be collected. Surgical evacuation will be performed if there is excessive bleeding due to incomplete abortion. Control visit
Study subjects will be requested to return for a control visit, two to three weeks after treatment at which a pelvic examination and ultrasonography will be performed. If the subject is found to have a continuing pregnancy, surgical evacuation will be performed. The results of all the tests relevant for the on-going care of the woman will be available to the treating physician.
3.3.7 Assessment of outcome of treatment The primary outcome of the study will be to ascertain the bioequivalence of the two formulations of misoprostol following sublingual administration. This is determined by the measurement of the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum concentration (tmax) and area under the curve (AUC) in each study group. The secondary outcome will be the efficacy (defined as the proportion of complete abortions in each study group) and side effects of each of the two product regimens. The complete abortion rate and the induction to abortion interval will also be compared. Adverse events, if any, will be analyzed on an intention to treat basis.
3.3.8 Criteria for discontinuation Of individual subjects Individual subjects will be discontinued if the woman:
• develops severe abdominal pain and heavy vaginal bleeding, at which point the pregnancy
will be terminated immediately by surgical evacuation. Heavy vaginal bleeding presenting an immediate threat to the patient’s health will be based on the judgement of the attending physician
Of the study If two or more subjects develop the same symptoms requiring discontinuation of the treatment and the principal investigator has good reason to believe and/or evidence to show that this (these) symptom(s) is (are) treatment-related, further recruitment will be suspended. The decision whether or not the study should be discontinued for this reason will be taken after discussion between the principal investigator and the Concept Foundation
Misoprostol acid The samples will be stored at below -20ºC and then sent in three batches to AAI Pharma GmbH & Co. KG laboratories (Neu-Ulm, Germany) for assay. Misoprostol acid (MPA) will be determined using liquid chromatography/tandem mass spectrometry (LC/MS/MS). The method is based on that of Yu Zou et al. (2007). Following liquid–liquid extraction, the analytes are separated using an isocratic mobile phase on a C18 column. A tandem mass spectrometer with an ionization source is used as the detector and operated in the negative ion mode. Multiple reaction monitoring using the precursor to product ion combinations of m/z 367–249 and 296–269 is performed to quantify misoprostol acid and the internal standard hydrochlorothiazide, respectively. The method is linear in the concentration range, 10.0–3000 pg/ml using 200μl of serum. The lower limit of quantification is 10.0 pg/ml. 3.3.10 Data management and analysis The serum levels of misoprostol acid will be recorded for each time interval and the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum
concentration (tmax) and area under the curve (AUC) will be calculated for each subject. Data analysis will be done as recommended by the EMEA for bioequivalence studies (EMEA, 2002). The statistical method for testing bioequivalence is based upon the 90% confidence interval for the ratio of the population means (test/reference), for the parameters under consideration. This method is equivalent to the corresponding two one-sided test procedure with the null hypothesis of bioequivalence at the 5% significance level (EMEA, 2002). Pharmacokinetic parameters derived from measures of concentration, eg AUC, Cmax will be analyzed using ANOVA. Prior to analysis, the data will be transformed using a logarithmic transformation. Tmax will also be calculated but this analysis will be non-parametric and applied to untransformed data. For all pharmacokinetic parameters, in addition to the appropriate 90% confidence intervals for the comparison of the two formulations, summary statistics such as median, minimum and maximum will be given.
3.3.11 Number of subjects European Medicines Agency recommendations for bioequivalence studies (EMEA, 2002), section 3.6.1 Statistical analysis, state: 'The statistical method for testing bioavailability (e.g. bioequivalence) is based upon the 90% confidence interval for the ratio of the population means (Test/Reference) for the parameters under consideration.' … For the AUC-ratio, 'the 90% confidence interval… should lie within an acceptance interval of 0.80-1.25.' It is also indicated that 'the data should be transformed prior to analysis using a logarithmic transformation.” Therefore, for bioequivalence, 0.80 < Test mean/Reference mean < 1.25 or, in the logarithmic scale, -0.22 = ln (0.80) < ln (Test mean) – ln (Reference mean) < 0.22. Bioequivalence will be demonstrated if the 90% confidence interval for the difference between means in the logarithmic scale is contained in -0.22-0.22 (margin of bioequivalence is 0.22). To obtain the SD in the transformed scale, the following approximate formula was used: Var(g(x)) = [dg(x)/dx]2 Var(x) = [dln(x)/dx]2 Var(x) = (1/x)2 Var(x), so that SD[ln(x)] ≈ SD(x)/x. A standard formula for sample size calculation for equivalence was used (Jones B et al, 1996). Misoprostol: From a study conducted assess the administration of misoprostol (Tang et al, 2002), the AUC for the vaginal route for 10 women had a mean of 433 pg.h/ml with a SD of 182. SD = 182/433 = 0.42 on a logarithmic scale. To obtain 80% statistical power with a 2-sided confidence interval of 90%, approximately 63 women would be needed in each arm to establish bioequivalence of the two misoprostol products within a margin of 0.22 in the logarithmic scale, which corresponds to the 90% confidence interval for the AUC-ratio within the interval of 0.80-1.25. The power obtained with 30 subjects in each arm will be 52%.
While 30 subjects/arm is in line with the recently completed pharmacokinetic study comparing the generic formulations of mifepristone administered orally and misoprostol administered vaginally with their original counterparts, it is uncertain whether it sufficient for assessing the bioequivalence of two formulations of misoprostol following sublingual administration. It is therefore proposed to recruit a total of 60 women (30 subjects/arm) and undertake an interim analysis. Should the data indicate that an adequate power has been achieved to ascertain the bioequivalence of the two formulations of each drug, the study will be terminated. If not, based on the data obtained, the number of additional women required will be calculated and the study continued to achieve this.
3.3.12 Duration of project The trial will require about 6 months for sample collection from the initial 60 women and a further 2 months to complete the assays of misoprostol and data analysis. It will be extended if determined to be necessary from the interim analysis.
3.4 Assessment of safety 3.4.1 Side-effects The use of mifepristone and misoprostol is associated with documented side-effects which have a causal relationship with the treatment. These side-effects include nausea, vomiting, diarrhoea, fatigue, dizziness, fainting, headache, lower abdominal pain or cramps, rash and chill or shivering. They usually occur within the first two hours after administration of misoprostol and, except for pain, are of low intensity. Side-effects will be recorded on the CRFs. 3.4.2 Adverse events Adverse events An adverse event is any untoward medical occurrence in a study subject administered a pharmaceutical product. It does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product. Serious adverse events A serious adverse event is any untoward medical occurrence that: - results in death - is life-threatening - requires in-patient hospitalization or prolongation of existing hospitalization - results in persistent or significant disability/incapacity - results in a congenital anomaly/birth defect Generally, inpatient hospitalization must include an overnight admission. Pre-planned elective procedures are not to be reported as serious adverse events. In this study continuing of pregnancy is not recorded as adverse event. It is considered as a lack of efficacy of the investigational product. 3.4.3 Procedures for reporting and recording adverse events The study subject will be given the opportunity to report adverse events spontaneously. A general prompt will also be given to detect adverse events. “Did you notice anything unusual about your health since last visit? All adverse events will be recorded using a standard adverse event form. In case of serious adverse events, the serious adverse event form will be used. The investigator will complete the initial report as soon as she/he receives the first information about the serious adverse event. The investigator forwards the serious adverse event form to Concept Foundation, Geneva by fax (attn: Peter Hall, +41-21-801 0109 and a scanned form to Helena von Hertzen (helena.vonhertzen@conceptfoundation.org). A serious adverse event will be followed up until it has resolved; has a stable level of sequelae; or the investigator no longer feels it is clinically significant. Concept Foundation and the Principal Investigator will communicate safety information to the Finnish Drug Regulatory Agency, NAM, according to applicable regulatory requirements. 3.5 Study management and administration
3.5.1 Compliance with good clinical practice (GCP) and good laboratory practice (GLP) The study has been designed to comply with the European Medicines Agency “Note for Guidance on Good Clinical Practice”, CPMP/ICH/135/95, July 2002. Standard operating procedures (SOPs) have been developed and arrangements made for their implementation to ensure that the study is conducted and that data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirements. Similarly, the laboratory analyses to be undertaken in the study will comply with Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the inspection and verification of good laboratory practice (GLP). 3.5.2 Monitoring The monitoring of the study is the responsibility of Concept Foundation. The monitor (person responsible for monitoring) will assist the Principal Investigator with the practical conduct of the study and assist her/him in working according to the protocol, GCP and regulatory requirements. The Principal Investigator will allow the sponsor or its representative to periodically monitor at mutually convenient times during and after the study. 3.5.3 Audit and inspection
The Principal Investigator will permit study-related audits by auditors mandated by Concept Foundation and inspections by domestic regulatory authorities, given reasonable notice. The main purposes of an audit or inspection are to confirm that the rights and well-being of the study subjects enrolled have been protected, and that all data relevant for evaluation of the investigational product have been processed and reported in compliance with the planned arrangements, GCP and applicable regulatory requirements. The investigator will provide direct access to all study documents. 3.5.4 Case Report Forms (CRF) The CRF is essentially a data entry form but will also constitute the original (source data) medical record as documented before the start of the study. 3.5.5 Investigator site file The content of the investigator site file has been structured in a manner that facilitates filing, retrieval, and/or auditing of study-related documents. 3.5.6 Product handling and accountability
The Head Pharmacist is responsible for receiving the study drugs at the Helsinki University Central Hospital pharmacy. The study drugs will be delivered via courier from Finnish drug wholesalers (Mifegyne®, Exelgyn and Cytotec®, Pfizer) and from Medikalla Oy - MediPharmia (generic mifepristone and misoprostol, Medabon®, Sun Pharma). The Head Pharmacist, Helsinki University Central Hospital will check the condition of the delivery and confirm its arrival and condition as requested by the supplier. If there is a problem with the delivered product, the supplier will be contacted for further instructions. The study drugs will be stored at the clinical trials unit in the Helsinki University Central Hospital pharmacy. The drugs will be stored at room temperature (<25°C) in a locked cabinet. The temperature is measured every 15 minutes by a calibrated electronic recorder and monitored daily. If the temperature increases above 25°C for more than 24h, Concept Foundation will be contacted for further instructions. The study drugs will be packed and labelled in the pharmacy in conformance with international
standards and according to a random table generated by the Head Pharmacist. The study drugs will be ordered from the ward by the study nurse via fax. These orders are to be signed by the principal investigator or, in his absence, the assistant investigator. The Head Pharmacist will pack the required order in a labelled paper bag, which will be sent from the pharmacy by car to the ward. Car deliveries are made routinely twice a week. On the ward, the study drugs will be stored in a locked cabinet reserved solely for them. The drugs will be stored at <25°C and the temperature monitored daily by the study nurse. The study nurse will record the receipt of the drugs on the Drug Accountability Form, as well as removal for administration to the study subjects. On completion of the study all unused study drugs, as well as empty packaging of the drugs used will be returned to the pharmacy. Any remaining drugs will be destroyed at the pharmacy following approval by Concept Foundation.
3.5.7 Data handling Concept Foundation will be responsible for data processing and analysis. 3.5.8 Clinical study report A clinical study report, conforming to the relevant ICH guidelines, will be prepared by The Concept Foundation together with the Principal Investigator.
3.7 Links with other projects: Nil 3.8 Expected study outcome It is expected that, following sublingual administration, the new generic misoprostol (co-packaged with mifepristone) product will be bioequivalent to the original formulation misoprostol. 3.9 References Ashok PW, Templeton A, Waagarachchi PT, Flett GM. (2002) Factors affecting the outcome of early medical abortion: a review of 4132 consecutive cases. Brit J Obstet Gynaecol 109:1281-1289
El-Refaey H, Rajasekar D, Abdalla M, Calder L, Templeton A. (1995) Induction of abortion with mifepristone (RU486) and oral or vaginal misoprostol. New Eng J Med 332:983-987 European Medicines Agency (2002). Note for guidance on the investigation of bioavailability and bioequivalence. http://www.egagenerics.com/doc/emea_bioequiv-1401-98.pdf
Hamoda H, Ashok PW, Dow J, Flett GM, Templeton A. (2003) A pilot study of mifepristone in combination with sublingual or vaginal misoprostol for medical termination of pregnancy up to 63 days gestation. Contraception, 68:335-8 Hamoda H, Ashok PW, Flett GM, Templeton A. (2005) A randomised controlled trial of mifepristone in combination with misoprostol administered sublingually or vaginally for medical abortion up to 13 weeks of gestation. Brit J Obstet Gynaecol, 112:1102-8
RCOG, Royal College of Obstetricians and Gynaecologists. (2004) The care of women requesting induced abortion: Evidence-based guideline No7. London, RCOG Press Tang OS, Schweer H, Seyberth NW, Lee SWH, Ho PC. (2002) Pharmacokinetics of different routes of administration of misoprostol. Human Reprod 17:332-6
Tang OS, Chan CC, Ng EH, Lee SW, Ho PC. (2003) A prospective, randomized, placebo-controlled trial on the use of mifepristone with sublingual or vaginal misoprostol for medical abortions of less than 9 weeks gestation. Human Reprod, 18:2315-2318. World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (1993). Termination of pregnancy with reduced doses of mifepristone (RU486) in late first trimester abortion. Contraception 50:461-475 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2000). Comparison of two doses of mifepristone in combination misoprostol for early medical abortion; a randomized trial. Brit J Obstet Gynaecol 107:524-530 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2003) WHO multinational study of three misoprostol regimens after mifepristone for early medical abortion. I: Efficacy. Brit J Obstet Gynaecol 110:808-818
Yu Zou, Xiaoyan Chen, Bo Song, Dafang Zhong (2007). Determination of misoprostol acid in human plasma by liquid chromatography coupled to tandem mass spectrometry. J Chromatog B, in press (Available online 13 January 2007)
Note: Start on this page and continue additional sheets as necessary.
Approvals will be obtained from the Ethics Committee of Gynaecology and Obstetrics, Otology, Ophtalmology, Neurology and Neurosurgery of the Hospital District of Helsinki and Uusimaa; and from the Finnish National Agency for Medicines. The study will be performed according to the Declaration of Helsinki.
All potential volunteers will be informed about the aims and protocol of the study, the side-effects that may occur as a result of the treatment. The measures taken to ensure confidentiality and their right to withdraw from the study at any time without prejudice to their further medical care will also be explained to them. Women participating in the study will be asked to sign the consent form.
Experience gained so far with the use of mifepristone followed by misoprostol for the termination of early pregnancy indicates that the therapy has side effects and a frequency of short-term complications comparable to those observed after vacuum aspiration. The most common complaint during treatment is lower abdominal pain, which results from uterine contractions and the abortion process itself – this is felt by practically all women. Using misoprostol, pregnancy-related symptoms, such as breast tenderness, nausea and vomiting decrease significantly during treatment and some women experience diarrhoea or shivering/fever during treatment. Less frequent side effects include dizziness, headache and rash. Side effects usually occur within the first two hours after misoprostol and are of low intensity, except for pain, which requires treatment in about half the cases.
Subjects will be informed about possible complications and qualified personnel and facilities will be available at all times to deal with them.
Most women participating in the study can be expected to have a complete abortion and
therefore will not be exposed to risks associated with vacuum aspiration, particularly the risks of physical trauma (cervical laceration, uterine perforation, etc). Even if surgical intervention is required in a small number of subjects, it is likely to be easier since the treatment will have dilated and softened the cervical canal.
No financial incentives will be offered to potential study participants and no financial remuneration will be given to women recruited into the study, other than reimbursement of expenses incurred as a result of participating in the study (travel expenses for extra hospital visits; lost days of work; and outpatient clinic fees). The subjects will be provided with a clinical follow-up, 2-3 weeks after the abortion.
Note: Ensure all information requested in the corresponding section of Part 1 is provided
1. Describe how the research addresses a demonstrated public health need and a need
Evidence that the new product is bioequivalent to the original products used for medical
abortion is essential to ensure its availability to many women as a choice of method of abortion. Sublingual administration of misoprostol will provide an alternative choice to women who do wish to undergo vaginal insertion of the tablets. This maybe a more acceptable option to some women. Furthermore, the availability of the two products in the most commonly use effective dose provided with appropriate information, will give additional reassurance that the drugs are provided by the physician at the correct dosage and at the right time.
2. How will the research reduce (or not increase) inequities in health and health care and
Poorer women will have access to choice in methods of abortion, something that in many
countries is only available to women who can afford private services.
3. Describe plans for disseminating results and sharing knowledge with research subjects
The results of the research will be communicated to the research subjects once the study is completed. They will be also communicated to the research community via scientific publication(s).
4. What is the sex composition of the research team? Describe whether the research topic
warrants female or male researchers specifically.
The principal investigator is male; the co-investigator and the research nurses are female.
The research project does not warrant female or male researchers specifically.
(Use extra pages, as necessary) 5a Budget
HUCH, Helsinki Staff and study costs
5b Timeline
NAM Clinical Laboratory Meetings Analysis (Use extra pages, as necessary)
5c Budget justification Briefly relate each item in the budget (personnel, supplies, subject costs, equipment, animals, etc.) to the activities outlined in the research proposal Senior Research Nurse A full-time research nurse is required for 6 months, to act as the main co-ordinator on a day-to-day basis under the direct supervision of the investigator. The nurse will be responsible for the recruitment of women, administration of mifepristone orally and of sublingual misoprostol. She will be responsible for blood sampling according to the schedule. She will also be responsible for the data entry. Research Nurse Because of the intensive blood sampling schedule, a single research nurse can undertake the recruitment of two subjects a week. The recruitment of a part-time (50%) research nurse will allow this to be increased to three subjects a week. The nurse will be responsible to the senior research nurse and will provide any necessary support for the recruitment of women, administration of drugs, blood sampling and data entry. Misoprostol acid assay The serum samples will now be sent to AAI Pharma GmbH & Co. KG laboratories (Neu-Ulm, Germany) for the assay of misoprostol acid. AAI Pharma GmbH & Co. KG laboratories is a GLP-accredited laboratory, and they are responsible for the establishment and validation of a modern LC/MS/MS assay for misoprostol acid and the subsequent analyses of samples from the study.
Form 6. Other support for the proposed research
(a) Is this research currently being supported directly by any other body?
If yes, give the name(s) of the Organization(s), nature, amount, and duration of support being provided
Concept will also be responsible for funding independent audit of good clinical practice (GCP) and of good laboratory practice (GLP) by South Barn Consultants Ltd, UK; and study monitoring and quality assessment by Medfiles, Medikalla OY, Finland; It will also be responsible for the validation of the misoprostol acid assay and the analyses of misoprostol acid by AAI Pharma GmbH & Co. KG.
(b) Is this application currently being considered elsewhere?
Form 7. Curricula vitae of the principal investigator(s) and co-investigator(s)
(Other formats containing the same information are acceptable.)
Please provide one form for each scientist involved in the project. Mikko A. Oskari Heikinheimo
Birth Current position
Associate professor / Dept of Ob&Gyn, University of Helsinki Senior physician / Hospital district of Helsinki and Uusimaa (HUS)
Senior lecturer (Ob&Gyn) / University of Helsinki
Training 1989
Jones Institute, EVMS, Norfolk, VA Research fellow / Reproductive endocrinology
Specialist in obstetrics and gynecology
Acting professor (obstetrics and gynecology)
Senior lecturer (obstetrics and gynecology)
Elected and additional positions 1997-2004
Promotion 2000 -committee/Univ of Helsinki
International asoprisnil advisory board /
Termination of pregnancy - committee for
Publications Research grants (>10.000 €) 1992
Sigrid Jusélius foundation – fellowship-grant
Sigrid Jusélius foundation – fellowship-grant
Finnish Medical Foundation – clinical investigator’s grant 150.000 €
Seth Wichmann award / Finnish society of obstetrics and gynecology, 2000
Official opponent
Riitta Koivunen, M.D. / University of Oulu, 24.8.2001
Dr. José Inzunza / Karolinska Institutet, Stockholm, 28.5.2003
Dr. Jennifer E. Scott / Karolinska Institutet, Stockholm, 7.4.2004
Tiina Backman, M.D. / University of Turku, 23.3.2007
Thesis reviewer
Annaleena Heikkilä, M.D. / University of Kuopio, 23.5.2003
Sirpa Hirvonen-Santti, M.D. / University of Helsinki, 10.12.2003
Marjut Otala, Ph.D. / University Helsinki, 17.12.2004
Med Dr. Christian Fiala / Karolinska Institutet, Stockholm, 7.10.2005
Stefan Karlsson, Ph.D. / Åbo Academi, Turku, 12.6.2006
Thesis supervisions
Helena Honkanen, M.D. / University of Helsinki, 16.1.2004 ’Medical abortion’ Päivi Lehtovirta, M.D. / University of Helsinki, 12.10.2007 ‘HIV in Ob&Gyn’ Selected publications 1996-2007
Heikinheimo O, Gordon K, Williams R, Hodgen G. Inhibition of ovulation by progestin analogs (agonists vs antagonists): preliminary evidence for different sites and mechanisms of actions. Contraception, 1996; 53: 55- 64. Heikinheimo O, Hsiu JG, Gordon K, Kim S, Williams R, Gibbons W, Hodgen G. Endometrial effects of RU486 - antiproliferative effects despite signs of estrogen action and increased cyclin-B expression. J Steroid Biochem Mol Biol, 1996; 59: 179-190. Greb R, Heikinheimo O, Williams R, Hodgen G, Goodman A. Vascular endothelial growth factor (VEGF) in primate endometrium is regulated by estrogen-receptor and progesterone-receptor ligands in vivo. Human Reproduction, 1997; 12: 1280-1292. Heikinheimo O, Ranta S, Moo-Young A, Lähteenmäki P, Gordon K. Parenteral administration of progestin Nestorone® to lactating cynomolgus monkeys: an ideal hormonal contraceptive at lactation? Human Reproduction, 1999; 14: 1993-1997. Hashiba Y, Asada Y, Heikinheimo O, Lanzendorf S, Mizutani S. Microinjection of antisense c-mos oligonucleotides prevents the progression of meiosis in human and hamster oocytes. Fertility and Sterility, 2001; 76: 143-147. Honkanen H, Ranta S, Ylikorkala O, Heikinheimo O. The kinetics of serum hCG and progesterone in response to oral and vaginal administration of misoprostol during medical termination of early pregnancy. Human Reproduction, 2002; 17: 2315-2319. Heikinheimo O, Raivio T, Honkanen H, Ranta S, Jänne OA. Termination of pregnancy with mifepristone and prostaglandin suppresses transiently circulating glucocorticoid bioactivity. J Clin Endocrinol Metab, 2003;88: 323-326. Honkanen H, Rutanen E-M, Heikinheimo O. Differential kinetics of serum and cervical insulin-like growth factor- binding protein-1 during mifepristone-misoprostol-induced medical termination of early pregnancy. Molecular Human Reproduction, 2004; 10: 65-70. Leminen R, Raivio T, Ranta S, Oehler J, von Hertzen H, Jänne AO, Heikinheimo O. Late follicular phase administration of mifepristone suppresses circulating leptin and follicle-stimulating hormone – mechanism(s) of action in emergency contraception? European Journal of Endocrinology, 2005; 152: 411-418. Heikinheimo O, Leminen R, Raivio T. Mifepristone in inhibition of the midcycle gonadotropin surge – evidence for both ovarian and pituitary sites of action. Fertility and Sterility, 2005, 84: 1545-1546. Heikinheimo O, Lehtovirta P, Suni J, Paavonen J. The levonorgestrel-releasing intrauterine system (LNG-IUS) in HIV-infected women – effects on bleeding patterns, ovarian function and genital shedding of HIV. Human Reproduction, 2006; 21: 2857-2861.
Scyl a serrata (Mud Crab) Seed Production And Its Health Management • Researcher : Dr. Lee Seong Wei • Co- Researcher : Dr. Ikhwanuddin b. Abdullah@Polity (UMT) • Studen : Cik Syahrizawati Binti Mohd Zohri • Grant : Short Term Research Grant Abstract The culture of mud crab, Scyl a spp , is widespread because the methods variety of farming technique exists for mud cr
Development in National Competition Laws Herausgeber:Bernhard Heitzer ´ Lioba Jüttner-Kramny ´ Karl M. MeessenLars-Hendrik Röller ´ Dirk SchroederEhrenmitglied: Alfred-Carl GaedertzDevelopments in National Competition Laws(January 1 ± March 31, 2009)This is the sixtieth quarterly report on developments in national competition laws publis-hed by Wirtschaft und Wettbewerb-WuW. This repo
 Particulars of the project and investigator(s)
Project No.:A65037-2 EUDRACT No.: 2008-003860-21
Particulars of the project and investigator(s)
Project No.:A65037-2 EUDRACT No.: 2008-003860-21
 • a history or evidence of thrombo-embolism, severe or recurrent liver disease or pruritus of
• has any medical condition or disease that requires regular treatment with systemic drugs,
care or precaution in conjunction with abortion
• tendency of abnormal bleeding (such as von Willebrandt’s disease)
• previous surgery of uterus/uterine cervix is a relative contraindication, however, previous
low-segment caesarean section is not a contraindication
• suspicion of any pathology of pregnancy (eg, molar, non-viable pregnancy, threatened
• suspected or known breast or genital neoplasia
• where difficulties are anticipated in follow-up.
Women older than 35 years, who smoke less than 20 cigarettes per day, have diastolic pressure of less than 90 mmHg and who have no known risk factor for cardiovascular disease, can be recruited into the study.
In addition, subjects who have been admitted into the study, will be excluded from the final
• the use of drugs, other than those prescribed by the investigator for the treatment of
possible therapy-related side-effects. Pain will be managed with premedication of 1g paracetamol and 30mg codeine, if required, NSAIDs or opiates will be given for additional pain medication
• essential data missing from the subject’s records making it impossible to judge the
• the presence of an acute illness of any nature during the treatment period.
The subjects will be randomized by computer-generated random numbers into two groups. The
random table will be generated by the Head Pharmacist, Helsinki University Central Hospital. Group A will comprise women who will be given 200mg of mifepristone produced by Exelgyn, followed 24 hours later by two doses of 2 tablets of 200μg misoprostol produced by Pfizer. Group B will comprise women who will be given 200mg of mifepristone and two doses of 2 tablets of 200μg misoprostol produced by Sun Pharma.
3.3.5 Description of the drugs to be used International Non-proprietary Name: Mifepristone (Mifegyne, Exelgyn and Medabon, Sun Pharma) Chemical name: 11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl) estra-4,9-dien-3-one
Route of administration: oral Amount: 1 x 200mg tablet International Non-proprietary Name: Misoprostol (Cytotec, Pfizer and Medabon, Sun Pharma)
• a history or evidence of thrombo-embolism, severe or recurrent liver disease or pruritus of
• has any medical condition or disease that requires regular treatment with systemic drugs,
care or precaution in conjunction with abortion
• tendency of abnormal bleeding (such as von Willebrandt’s disease)
• previous surgery of uterus/uterine cervix is a relative contraindication, however, previous
low-segment caesarean section is not a contraindication
• suspicion of any pathology of pregnancy (eg, molar, non-viable pregnancy, threatened
• suspected or known breast or genital neoplasia
• where difficulties are anticipated in follow-up.
Women older than 35 years, who smoke less than 20 cigarettes per day, have diastolic pressure of less than 90 mmHg and who have no known risk factor for cardiovascular disease, can be recruited into the study.
In addition, subjects who have been admitted into the study, will be excluded from the final
• the use of drugs, other than those prescribed by the investigator for the treatment of
possible therapy-related side-effects. Pain will be managed with premedication of 1g paracetamol and 30mg codeine, if required, NSAIDs or opiates will be given for additional pain medication
• essential data missing from the subject’s records making it impossible to judge the
• the presence of an acute illness of any nature during the treatment period.
The subjects will be randomized by computer-generated random numbers into two groups. The
random table will be generated by the Head Pharmacist, Helsinki University Central Hospital. Group A will comprise women who will be given 200mg of mifepristone produced by Exelgyn, followed 24 hours later by two doses of 2 tablets of 200μg misoprostol produced by Pfizer. Group B will comprise women who will be given 200mg of mifepristone and two doses of 2 tablets of 200μg misoprostol produced by Sun Pharma.
3.3.5 Description of the drugs to be used International Non-proprietary Name: Mifepristone (Mifegyne, Exelgyn and Medabon, Sun Pharma) Chemical name: 11β-[p-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl) estra-4,9-dien-3-one
Route of administration: oral Amount: 1 x 200mg tablet International Non-proprietary Name: Misoprostol (Cytotec, Pfizer and Medabon, Sun Pharma)
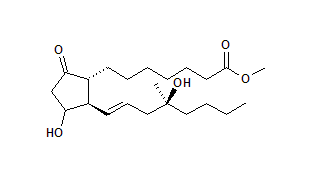 Chemical name: 7-[3-hydroxy-2-(4-hydroxy-4-methyl-oct-1-enyl)-5-oxo-cyclopentyl] heptanoic acid
Route of administration: sublingual Amount: 4 x 200 μg tablets
3.3.6 Admission procedure, treatment and sampling schedule
Before admission to the study, potential subjects will undergo the following:
• ultrasound examination to verify the length of the pregnancy and check that the pregnancy
is intrauterine. If the pregnancy is more advanced, if the there is a suspicion of an abnormal pregnancy (eg, extra-uterine, molar, etc) the woman will not be admitted into the study
• medical, obstetrical and gynaecological history
• medical and gynaecological examination
Women who fulfil the admission criteria, are willing and able to participate in the study and have given their signed informed consent will be included in the study. The study will be performed over a period of two days during the medical termination of pregnancy. Day one Baseline blood pressure, pulse rate, body height and weight will be taken. Medical and obstetrical history, as well as informed consent, will be obtained. The subjects will be randomized by computer-generated random numbers into 2 groups. Each woman will then be given 200mg of mifepristone orally. Following ingestion of mifepristone women are free to leave the hospital. Day two (24h after oral administration of mifepristone) An intravenous catheter suitable for multiple blood sampling will be inserted. A venous blood sample (10ml) will be collected before misoprostol administration; this will be the 0min sample prior to misoprostol. Two tablets of 200μg misoprostol will then be administered sublingually. After 20 minutes the tablets can be swallowed if they have not melted and another two tablets will be administered sublingually. Venous blood samples (10ml) will be taken at 15, 30, 45, 60, 75, 90 and 105min and 2, 3, 4 and 6 hours after administration of misoprostol. All blood samples will be centrifuged without delay and serum will be immediately frozen and stored at -20ºC. Blood pressure, pulse and any reported side effects will be monitored. The interval from the administration of misoprostol to onset of abdominal pain/vaginal bleeding and passage of production of conception will be recorded. The research nurse will stay with the subjects throughout the blood taking procedure to provide emotional support and counselling if necessary. It is anticipated that some 90% of subjects will have aborted by six hours after the administration of misoprostol. Some subjects will abort before six hours, in which case no further blood samples will be collected. Surgical evacuation will be performed if there is excessive bleeding due to incomplete abortion. Control visit
Study subjects will be requested to return for a control visit, two to three weeks after treatment at which a pelvic examination and ultrasonography will be performed. If the subject is found to have a continuing pregnancy, surgical evacuation will be performed. The results of all the tests relevant for the on-going care of the woman will be available to the treating physician.
3.3.7 Assessment of outcome of treatment The primary outcome of the study will be to ascertain the bioequivalence of the two formulations of misoprostol following sublingual administration. This is determined by the measurement of the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum concentration (tmax) and area under the curve (AUC) in each study group. The secondary outcome will be the efficacy (defined as the proportion of complete abortions in each study group) and side effects of each of the two product regimens. The complete abortion rate and the induction to abortion interval will also be compared. Adverse events, if any, will be analyzed on an intention to treat basis.
3.3.8 Criteria for discontinuation Of individual subjects Individual subjects will be discontinued if the woman:
• develops severe abdominal pain and heavy vaginal bleeding, at which point the pregnancy
will be terminated immediately by surgical evacuation. Heavy vaginal bleeding presenting an immediate threat to the patient’s health will be based on the judgement of the attending physician
Of the study If two or more subjects develop the same symptoms requiring discontinuation of the treatment and the principal investigator has good reason to believe and/or evidence to show that this (these) symptom(s) is (are) treatment-related, further recruitment will be suspended. The decision whether or not the study should be discontinued for this reason will be taken after discussion between the principal investigator and the Concept Foundation
Misoprostol acid The samples will be stored at below -20ºC and then sent in three batches to AAI Pharma GmbH & Co. KG laboratories (Neu-Ulm, Germany) for assay. Misoprostol acid (MPA) will be determined using liquid chromatography/tandem mass spectrometry (LC/MS/MS). The method is based on that of Yu Zou et al. (2007). Following liquid–liquid extraction, the analytes are separated using an isocratic mobile phase on a C18 column. A tandem mass spectrometer with an ionization source is used as the detector and operated in the negative ion mode. Multiple reaction monitoring using the precursor to product ion combinations of m/z 367–249 and 296–269 is performed to quantify misoprostol acid and the internal standard hydrochlorothiazide, respectively. The method is linear in the concentration range, 10.0–3000 pg/ml using 200μl of serum. The lower limit of quantification is 10.0 pg/ml. 3.3.10 Data management and analysis The serum levels of misoprostol acid will be recorded for each time interval and the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum
concentration (tmax) and area under the curve (AUC) will be calculated for each subject. Data analysis will be done as recommended by the EMEA for bioequivalence studies (EMEA, 2002). The statistical method for testing bioequivalence is based upon the 90% confidence interval for the ratio of the population means (test/reference), for the parameters under consideration. This method is equivalent to the corresponding two one-sided test procedure with the null hypothesis of bioequivalence at the 5% significance level (EMEA, 2002). Pharmacokinetic parameters derived from measures of concentration, eg AUC, Cmax will be analyzed using ANOVA. Prior to analysis, the data will be transformed using a logarithmic transformation. Tmax will also be calculated but this analysis will be non-parametric and applied to untransformed data. For all pharmacokinetic parameters, in addition to the appropriate 90% confidence intervals for the comparison of the two formulations, summary statistics such as median, minimum and maximum will be given.
3.3.11 Number of subjects European Medicines Agency recommendations for bioequivalence studies (EMEA, 2002), section 3.6.1 Statistical analysis, state: 'The statistical method for testing bioavailability (e.g. bioequivalence) is based upon the 90% confidence interval for the ratio of the population means (Test/Reference) for the parameters under consideration.' … For the AUC-ratio, 'the 90% confidence interval… should lie within an acceptance interval of 0.80-1.25.' It is also indicated that 'the data should be transformed prior to analysis using a logarithmic transformation.” Therefore, for bioequivalence, 0.80 < Test mean/Reference mean < 1.25 or, in the logarithmic scale, -0.22 = ln (0.80) < ln (Test mean) – ln (Reference mean) < 0.22. Bioequivalence will be demonstrated if the 90% confidence interval for the difference between means in the logarithmic scale is contained in -0.22-0.22 (margin of bioequivalence is 0.22). To obtain the SD in the transformed scale, the following approximate formula was used: Var(g(x)) = [dg(x)/dx]2 Var(x) = [dln(x)/dx]2 Var(x) = (1/x)2 Var(x), so that SD[ln(x)] ≈ SD(x)/x. A standard formula for sample size calculation for equivalence was used (Jones B et al, 1996). Misoprostol: From a study conducted assess the administration of misoprostol (Tang et al, 2002), the AUC for the vaginal route for 10 women had a mean of 433 pg.h/ml with a SD of 182. SD = 182/433 = 0.42 on a logarithmic scale. To obtain 80% statistical power with a 2-sided confidence interval of 90%, approximately 63 women would be needed in each arm to establish bioequivalence of the two misoprostol products within a margin of 0.22 in the logarithmic scale, which corresponds to the 90% confidence interval for the AUC-ratio within the interval of 0.80-1.25. The power obtained with 30 subjects in each arm will be 52%.
While 30 subjects/arm is in line with the recently completed pharmacokinetic study comparing the generic formulations of mifepristone administered orally and misoprostol administered vaginally with their original counterparts, it is uncertain whether it sufficient for assessing the bioequivalence of two formulations of misoprostol following sublingual administration. It is therefore proposed to recruit a total of 60 women (30 subjects/arm) and undertake an interim analysis. Should the data indicate that an adequate power has been achieved to ascertain the bioequivalence of the two formulations of each drug, the study will be terminated. If not, based on the data obtained, the number of additional women required will be calculated and the study continued to achieve this.
3.3.12 Duration of project The trial will require about 6 months for sample collection from the initial 60 women and a further 2 months to complete the assays of misoprostol and data analysis. It will be extended if determined to be necessary from the interim analysis.
3.4 Assessment of safety 3.4.1 Side-effects The use of mifepristone and misoprostol is associated with documented side-effects which have a causal relationship with the treatment. These side-effects include nausea, vomiting, diarrhoea, fatigue, dizziness, fainting, headache, lower abdominal pain or cramps, rash and chill or shivering. They usually occur within the first two hours after administration of misoprostol and, except for pain, are of low intensity. Side-effects will be recorded on the CRFs. 3.4.2 Adverse events Adverse events An adverse event is any untoward medical occurrence in a study subject administered a pharmaceutical product. It does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product. Serious adverse events A serious adverse event is any untoward medical occurrence that: - results in death - is life-threatening - requires in-patient hospitalization or prolongation of existing hospitalization - results in persistent or significant disability/incapacity - results in a congenital anomaly/birth defect Generally, inpatient hospitalization must include an overnight admission. Pre-planned elective procedures are not to be reported as serious adverse events. In this study continuing of pregnancy is not recorded as adverse event. It is considered as a lack of efficacy of the investigational product. 3.4.3 Procedures for reporting and recording adverse events The study subject will be given the opportunity to report adverse events spontaneously. A general prompt will also be given to detect adverse events. “Did you notice anything unusual about your health since last visit? All adverse events will be recorded using a standard adverse event form. In case of serious adverse events, the serious adverse event form will be used. The investigator will complete the initial report as soon as she/he receives the first information about the serious adverse event. The investigator forwards the serious adverse event form to Concept Foundation, Geneva by fax (attn: Peter Hall, +41-21-801 0109 and a scanned form to Helena von Hertzen (helena.vonhertzen@conceptfoundation.org). A serious adverse event will be followed up until it has resolved; has a stable level of sequelae; or the investigator no longer feels it is clinically significant. Concept Foundation and the Principal Investigator will communicate safety information to the Finnish Drug Regulatory Agency, NAM, according to applicable regulatory requirements. 3.5 Study management and administration
3.5.1 Compliance with good clinical practice (GCP) and good laboratory practice (GLP) The study has been designed to comply with the European Medicines Agency “Note for Guidance on Good Clinical Practice”, CPMP/ICH/135/95, July 2002. Standard operating procedures (SOPs) have been developed and arrangements made for their implementation to ensure that the study is conducted and that data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirements. Similarly, the laboratory analyses to be undertaken in the study will comply with Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the inspection and verification of good laboratory practice (GLP). 3.5.2 Monitoring The monitoring of the study is the responsibility of Concept Foundation. The monitor (person responsible for monitoring) will assist the Principal Investigator with the practical conduct of the study and assist her/him in working according to the protocol, GCP and regulatory requirements. The Principal Investigator will allow the sponsor or its representative to periodically monitor at mutually convenient times during and after the study. 3.5.3 Audit and inspection
The Principal Investigator will permit study-related audits by auditors mandated by Concept Foundation and inspections by domestic regulatory authorities, given reasonable notice. The main purposes of an audit or inspection are to confirm that the rights and well-being of the study subjects enrolled have been protected, and that all data relevant for evaluation of the investigational product have been processed and reported in compliance with the planned arrangements, GCP and applicable regulatory requirements. The investigator will provide direct access to all study documents. 3.5.4 Case Report Forms (CRF) The CRF is essentially a data entry form but will also constitute the original (source data) medical record as documented before the start of the study. 3.5.5 Investigator site file The content of the investigator site file has been structured in a manner that facilitates filing, retrieval, and/or auditing of study-related documents. 3.5.6 Product handling and accountability
The Head Pharmacist is responsible for receiving the study drugs at the Helsinki University Central Hospital pharmacy. The study drugs will be delivered via courier from Finnish drug wholesalers (Mifegyne®, Exelgyn and Cytotec®, Pfizer) and from Medikalla Oy - MediPharmia (generic mifepristone and misoprostol, Medabon®, Sun Pharma). The Head Pharmacist, Helsinki University Central Hospital will check the condition of the delivery and confirm its arrival and condition as requested by the supplier. If there is a problem with the delivered product, the supplier will be contacted for further instructions. The study drugs will be stored at the clinical trials unit in the Helsinki University Central Hospital pharmacy. The drugs will be stored at room temperature (<25°C) in a locked cabinet. The temperature is measured every 15 minutes by a calibrated electronic recorder and monitored daily. If the temperature increases above 25°C for more than 24h, Concept Foundation will be contacted for further instructions. The study drugs will be packed and labelled in the pharmacy in conformance with international
standards and according to a random table generated by the Head Pharmacist. The study drugs will be ordered from the ward by the study nurse via fax. These orders are to be signed by the principal investigator or, in his absence, the assistant investigator. The Head Pharmacist will pack the required order in a labelled paper bag, which will be sent from the pharmacy by car to the ward. Car deliveries are made routinely twice a week. On the ward, the study drugs will be stored in a locked cabinet reserved solely for them. The drugs will be stored at <25°C and the temperature monitored daily by the study nurse. The study nurse will record the receipt of the drugs on the Drug Accountability Form, as well as removal for administration to the study subjects. On completion of the study all unused study drugs, as well as empty packaging of the drugs used will be returned to the pharmacy. Any remaining drugs will be destroyed at the pharmacy following approval by Concept Foundation.
3.5.7 Data handling Concept Foundation will be responsible for data processing and analysis. 3.5.8 Clinical study report A clinical study report, conforming to the relevant ICH guidelines, will be prepared by The Concept Foundation together with the Principal Investigator.
3.7 Links with other projects: Nil 3.8 Expected study outcome It is expected that, following sublingual administration, the new generic misoprostol (co-packaged with mifepristone) product will be bioequivalent to the original formulation misoprostol. 3.9 References Ashok PW, Templeton A, Waagarachchi PT, Flett GM. (2002) Factors affecting the outcome of early medical abortion: a review of 4132 consecutive cases. Brit J Obstet Gynaecol 109:1281-1289
El-Refaey H, Rajasekar D, Abdalla M, Calder L, Templeton A. (1995) Induction of abortion with mifepristone (RU486) and oral or vaginal misoprostol. New Eng J Med 332:983-987 European Medicines Agency (2002). Note for guidance on the investigation of bioavailability and bioequivalence. http://www.egagenerics.com/doc/emea_bioequiv-1401-98.pdf
Hamoda H, Ashok PW, Dow J, Flett GM, Templeton A. (2003) A pilot study of mifepristone in combination with sublingual or vaginal misoprostol for medical termination of pregnancy up to 63 days gestation. Contraception, 68:335-8 Hamoda H, Ashok PW, Flett GM, Templeton A. (2005) A randomised controlled trial of mifepristone in combination with misoprostol administered sublingually or vaginally for medical abortion up to 13 weeks of gestation. Brit J Obstet Gynaecol, 112:1102-8
RCOG, Royal College of Obstetricians and Gynaecologists. (2004) The care of women requesting induced abortion: Evidence-based guideline No7. London, RCOG Press Tang OS, Schweer H, Seyberth NW, Lee SWH, Ho PC. (2002) Pharmacokinetics of different routes of administration of misoprostol. Human Reprod 17:332-6
Tang OS, Chan CC, Ng EH, Lee SW, Ho PC. (2003) A prospective, randomized, placebo-controlled trial on the use of mifepristone with sublingual or vaginal misoprostol for medical abortions of less than 9 weeks gestation. Human Reprod, 18:2315-2318. World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (1993). Termination of pregnancy with reduced doses of mifepristone (RU486) in late first trimester abortion. Contraception 50:461-475 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2000). Comparison of two doses of mifepristone in combination misoprostol for early medical abortion; a randomized trial. Brit J Obstet Gynaecol 107:524-530 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2003) WHO multinational study of three misoprostol regimens after mifepristone for early medical abortion. I: Efficacy. Brit J Obstet Gynaecol 110:808-818
Yu Zou, Xiaoyan Chen, Bo Song, Dafang Zhong (2007). Determination of misoprostol acid in human plasma by liquid chromatography coupled to tandem mass spectrometry. J Chromatog B, in press (Available online 13 January 2007)
Note: Start on this page and continue additional sheets as necessary.
Approvals will be obtained from the Ethics Committee of Gynaecology and Obstetrics, Otology, Ophtalmology, Neurology and Neurosurgery of the Hospital District of Helsinki and Uusimaa; and from the Finnish National Agency for Medicines. The study will be performed according to the Declaration of Helsinki.
All potential volunteers will be informed about the aims and protocol of the study, the side-effects that may occur as a result of the treatment. The measures taken to ensure confidentiality and their right to withdraw from the study at any time without prejudice to their further medical care will also be explained to them. Women participating in the study will be asked to sign the consent form.
Experience gained so far with the use of mifepristone followed by misoprostol for the termination of early pregnancy indicates that the therapy has side effects and a frequency of short-term complications comparable to those observed after vacuum aspiration. The most common complaint during treatment is lower abdominal pain, which results from uterine contractions and the abortion process itself – this is felt by practically all women. Using misoprostol, pregnancy-related symptoms, such as breast tenderness, nausea and vomiting decrease significantly during treatment and some women experience diarrhoea or shivering/fever during treatment. Less frequent side effects include dizziness, headache and rash. Side effects usually occur within the first two hours after misoprostol and are of low intensity, except for pain, which requires treatment in about half the cases.
Subjects will be informed about possible complications and qualified personnel and facilities will be available at all times to deal with them.
Most women participating in the study can be expected to have a complete abortion and
therefore will not be exposed to risks associated with vacuum aspiration, particularly the risks of physical trauma (cervical laceration, uterine perforation, etc). Even if surgical intervention is required in a small number of subjects, it is likely to be easier since the treatment will have dilated and softened the cervical canal.
No financial incentives will be offered to potential study participants and no financial remuneration will be given to women recruited into the study, other than reimbursement of expenses incurred as a result of participating in the study (travel expenses for extra hospital visits; lost days of work; and outpatient clinic fees). The subjects will be provided with a clinical follow-up, 2-3 weeks after the abortion.
Note: Ensure all information requested in the corresponding section of Part 1 is provided
Chemical name: 7-[3-hydroxy-2-(4-hydroxy-4-methyl-oct-1-enyl)-5-oxo-cyclopentyl] heptanoic acid
Route of administration: sublingual Amount: 4 x 200 μg tablets
3.3.6 Admission procedure, treatment and sampling schedule
Before admission to the study, potential subjects will undergo the following:
• ultrasound examination to verify the length of the pregnancy and check that the pregnancy
is intrauterine. If the pregnancy is more advanced, if the there is a suspicion of an abnormal pregnancy (eg, extra-uterine, molar, etc) the woman will not be admitted into the study
• medical, obstetrical and gynaecological history
• medical and gynaecological examination
Women who fulfil the admission criteria, are willing and able to participate in the study and have given their signed informed consent will be included in the study. The study will be performed over a period of two days during the medical termination of pregnancy. Day one Baseline blood pressure, pulse rate, body height and weight will be taken. Medical and obstetrical history, as well as informed consent, will be obtained. The subjects will be randomized by computer-generated random numbers into 2 groups. Each woman will then be given 200mg of mifepristone orally. Following ingestion of mifepristone women are free to leave the hospital. Day two (24h after oral administration of mifepristone) An intravenous catheter suitable for multiple blood sampling will be inserted. A venous blood sample (10ml) will be collected before misoprostol administration; this will be the 0min sample prior to misoprostol. Two tablets of 200μg misoprostol will then be administered sublingually. After 20 minutes the tablets can be swallowed if they have not melted and another two tablets will be administered sublingually. Venous blood samples (10ml) will be taken at 15, 30, 45, 60, 75, 90 and 105min and 2, 3, 4 and 6 hours after administration of misoprostol. All blood samples will be centrifuged without delay and serum will be immediately frozen and stored at -20ºC. Blood pressure, pulse and any reported side effects will be monitored. The interval from the administration of misoprostol to onset of abdominal pain/vaginal bleeding and passage of production of conception will be recorded. The research nurse will stay with the subjects throughout the blood taking procedure to provide emotional support and counselling if necessary. It is anticipated that some 90% of subjects will have aborted by six hours after the administration of misoprostol. Some subjects will abort before six hours, in which case no further blood samples will be collected. Surgical evacuation will be performed if there is excessive bleeding due to incomplete abortion. Control visit
Study subjects will be requested to return for a control visit, two to three weeks after treatment at which a pelvic examination and ultrasonography will be performed. If the subject is found to have a continuing pregnancy, surgical evacuation will be performed. The results of all the tests relevant for the on-going care of the woman will be available to the treating physician.
3.3.7 Assessment of outcome of treatment The primary outcome of the study will be to ascertain the bioequivalence of the two formulations of misoprostol following sublingual administration. This is determined by the measurement of the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum concentration (tmax) and area under the curve (AUC) in each study group. The secondary outcome will be the efficacy (defined as the proportion of complete abortions in each study group) and side effects of each of the two product regimens. The complete abortion rate and the induction to abortion interval will also be compared. Adverse events, if any, will be analyzed on an intention to treat basis.
3.3.8 Criteria for discontinuation Of individual subjects Individual subjects will be discontinued if the woman:
• develops severe abdominal pain and heavy vaginal bleeding, at which point the pregnancy
will be terminated immediately by surgical evacuation. Heavy vaginal bleeding presenting an immediate threat to the patient’s health will be based on the judgement of the attending physician
Of the study If two or more subjects develop the same symptoms requiring discontinuation of the treatment and the principal investigator has good reason to believe and/or evidence to show that this (these) symptom(s) is (are) treatment-related, further recruitment will be suspended. The decision whether or not the study should be discontinued for this reason will be taken after discussion between the principal investigator and the Concept Foundation
Misoprostol acid The samples will be stored at below -20ºC and then sent in three batches to AAI Pharma GmbH & Co. KG laboratories (Neu-Ulm, Germany) for assay. Misoprostol acid (MPA) will be determined using liquid chromatography/tandem mass spectrometry (LC/MS/MS). The method is based on that of Yu Zou et al. (2007). Following liquid–liquid extraction, the analytes are separated using an isocratic mobile phase on a C18 column. A tandem mass spectrometer with an ionization source is used as the detector and operated in the negative ion mode. Multiple reaction monitoring using the precursor to product ion combinations of m/z 367–249 and 296–269 is performed to quantify misoprostol acid and the internal standard hydrochlorothiazide, respectively. The method is linear in the concentration range, 10.0–3000 pg/ml using 200μl of serum. The lower limit of quantification is 10.0 pg/ml. 3.3.10 Data management and analysis The serum levels of misoprostol acid will be recorded for each time interval and the pharmacokinetic parameters, maximum serum concentration (Cmax), time to maximum serum
concentration (tmax) and area under the curve (AUC) will be calculated for each subject. Data analysis will be done as recommended by the EMEA for bioequivalence studies (EMEA, 2002). The statistical method for testing bioequivalence is based upon the 90% confidence interval for the ratio of the population means (test/reference), for the parameters under consideration. This method is equivalent to the corresponding two one-sided test procedure with the null hypothesis of bioequivalence at the 5% significance level (EMEA, 2002). Pharmacokinetic parameters derived from measures of concentration, eg AUC, Cmax will be analyzed using ANOVA. Prior to analysis, the data will be transformed using a logarithmic transformation. Tmax will also be calculated but this analysis will be non-parametric and applied to untransformed data. For all pharmacokinetic parameters, in addition to the appropriate 90% confidence intervals for the comparison of the two formulations, summary statistics such as median, minimum and maximum will be given.
3.3.11 Number of subjects European Medicines Agency recommendations for bioequivalence studies (EMEA, 2002), section 3.6.1 Statistical analysis, state: 'The statistical method for testing bioavailability (e.g. bioequivalence) is based upon the 90% confidence interval for the ratio of the population means (Test/Reference) for the parameters under consideration.' … For the AUC-ratio, 'the 90% confidence interval… should lie within an acceptance interval of 0.80-1.25.' It is also indicated that 'the data should be transformed prior to analysis using a logarithmic transformation.” Therefore, for bioequivalence, 0.80 < Test mean/Reference mean < 1.25 or, in the logarithmic scale, -0.22 = ln (0.80) < ln (Test mean) – ln (Reference mean) < 0.22. Bioequivalence will be demonstrated if the 90% confidence interval for the difference between means in the logarithmic scale is contained in -0.22-0.22 (margin of bioequivalence is 0.22). To obtain the SD in the transformed scale, the following approximate formula was used: Var(g(x)) = [dg(x)/dx]2 Var(x) = [dln(x)/dx]2 Var(x) = (1/x)2 Var(x), so that SD[ln(x)] ≈ SD(x)/x. A standard formula for sample size calculation for equivalence was used (Jones B et al, 1996). Misoprostol: From a study conducted assess the administration of misoprostol (Tang et al, 2002), the AUC for the vaginal route for 10 women had a mean of 433 pg.h/ml with a SD of 182. SD = 182/433 = 0.42 on a logarithmic scale. To obtain 80% statistical power with a 2-sided confidence interval of 90%, approximately 63 women would be needed in each arm to establish bioequivalence of the two misoprostol products within a margin of 0.22 in the logarithmic scale, which corresponds to the 90% confidence interval for the AUC-ratio within the interval of 0.80-1.25. The power obtained with 30 subjects in each arm will be 52%.
While 30 subjects/arm is in line with the recently completed pharmacokinetic study comparing the generic formulations of mifepristone administered orally and misoprostol administered vaginally with their original counterparts, it is uncertain whether it sufficient for assessing the bioequivalence of two formulations of misoprostol following sublingual administration. It is therefore proposed to recruit a total of 60 women (30 subjects/arm) and undertake an interim analysis. Should the data indicate that an adequate power has been achieved to ascertain the bioequivalence of the two formulations of each drug, the study will be terminated. If not, based on the data obtained, the number of additional women required will be calculated and the study continued to achieve this.
3.3.12 Duration of project The trial will require about 6 months for sample collection from the initial 60 women and a further 2 months to complete the assays of misoprostol and data analysis. It will be extended if determined to be necessary from the interim analysis.
3.4 Assessment of safety 3.4.1 Side-effects The use of mifepristone and misoprostol is associated with documented side-effects which have a causal relationship with the treatment. These side-effects include nausea, vomiting, diarrhoea, fatigue, dizziness, fainting, headache, lower abdominal pain or cramps, rash and chill or shivering. They usually occur within the first two hours after administration of misoprostol and, except for pain, are of low intensity. Side-effects will be recorded on the CRFs. 3.4.2 Adverse events Adverse events An adverse event is any untoward medical occurrence in a study subject administered a pharmaceutical product. It does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product. Serious adverse events A serious adverse event is any untoward medical occurrence that: - results in death - is life-threatening - requires in-patient hospitalization or prolongation of existing hospitalization - results in persistent or significant disability/incapacity - results in a congenital anomaly/birth defect Generally, inpatient hospitalization must include an overnight admission. Pre-planned elective procedures are not to be reported as serious adverse events. In this study continuing of pregnancy is not recorded as adverse event. It is considered as a lack of efficacy of the investigational product. 3.4.3 Procedures for reporting and recording adverse events The study subject will be given the opportunity to report adverse events spontaneously. A general prompt will also be given to detect adverse events. “Did you notice anything unusual about your health since last visit? All adverse events will be recorded using a standard adverse event form. In case of serious adverse events, the serious adverse event form will be used. The investigator will complete the initial report as soon as she/he receives the first information about the serious adverse event. The investigator forwards the serious adverse event form to Concept Foundation, Geneva by fax (attn: Peter Hall, +41-21-801 0109 and a scanned form to Helena von Hertzen (helena.vonhertzen@conceptfoundation.org). A serious adverse event will be followed up until it has resolved; has a stable level of sequelae; or the investigator no longer feels it is clinically significant. Concept Foundation and the Principal Investigator will communicate safety information to the Finnish Drug Regulatory Agency, NAM, according to applicable regulatory requirements. 3.5 Study management and administration
3.5.1 Compliance with good clinical practice (GCP) and good laboratory practice (GLP) The study has been designed to comply with the European Medicines Agency “Note for Guidance on Good Clinical Practice”, CPMP/ICH/135/95, July 2002. Standard operating procedures (SOPs) have been developed and arrangements made for their implementation to ensure that the study is conducted and that data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirements. Similarly, the laboratory analyses to be undertaken in the study will comply with Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the inspection and verification of good laboratory practice (GLP). 3.5.2 Monitoring The monitoring of the study is the responsibility of Concept Foundation. The monitor (person responsible for monitoring) will assist the Principal Investigator with the practical conduct of the study and assist her/him in working according to the protocol, GCP and regulatory requirements. The Principal Investigator will allow the sponsor or its representative to periodically monitor at mutually convenient times during and after the study. 3.5.3 Audit and inspection
The Principal Investigator will permit study-related audits by auditors mandated by Concept Foundation and inspections by domestic regulatory authorities, given reasonable notice. The main purposes of an audit or inspection are to confirm that the rights and well-being of the study subjects enrolled have been protected, and that all data relevant for evaluation of the investigational product have been processed and reported in compliance with the planned arrangements, GCP and applicable regulatory requirements. The investigator will provide direct access to all study documents. 3.5.4 Case Report Forms (CRF) The CRF is essentially a data entry form but will also constitute the original (source data) medical record as documented before the start of the study. 3.5.5 Investigator site file The content of the investigator site file has been structured in a manner that facilitates filing, retrieval, and/or auditing of study-related documents. 3.5.6 Product handling and accountability
The Head Pharmacist is responsible for receiving the study drugs at the Helsinki University Central Hospital pharmacy. The study drugs will be delivered via courier from Finnish drug wholesalers (Mifegyne®, Exelgyn and Cytotec®, Pfizer) and from Medikalla Oy - MediPharmia (generic mifepristone and misoprostol, Medabon®, Sun Pharma). The Head Pharmacist, Helsinki University Central Hospital will check the condition of the delivery and confirm its arrival and condition as requested by the supplier. If there is a problem with the delivered product, the supplier will be contacted for further instructions. The study drugs will be stored at the clinical trials unit in the Helsinki University Central Hospital pharmacy. The drugs will be stored at room temperature (<25°C) in a locked cabinet. The temperature is measured every 15 minutes by a calibrated electronic recorder and monitored daily. If the temperature increases above 25°C for more than 24h, Concept Foundation will be contacted for further instructions. The study drugs will be packed and labelled in the pharmacy in conformance with international
standards and according to a random table generated by the Head Pharmacist. The study drugs will be ordered from the ward by the study nurse via fax. These orders are to be signed by the principal investigator or, in his absence, the assistant investigator. The Head Pharmacist will pack the required order in a labelled paper bag, which will be sent from the pharmacy by car to the ward. Car deliveries are made routinely twice a week. On the ward, the study drugs will be stored in a locked cabinet reserved solely for them. The drugs will be stored at <25°C and the temperature monitored daily by the study nurse. The study nurse will record the receipt of the drugs on the Drug Accountability Form, as well as removal for administration to the study subjects. On completion of the study all unused study drugs, as well as empty packaging of the drugs used will be returned to the pharmacy. Any remaining drugs will be destroyed at the pharmacy following approval by Concept Foundation.
3.5.7 Data handling Concept Foundation will be responsible for data processing and analysis. 3.5.8 Clinical study report A clinical study report, conforming to the relevant ICH guidelines, will be prepared by The Concept Foundation together with the Principal Investigator.
3.7 Links with other projects: Nil 3.8 Expected study outcome It is expected that, following sublingual administration, the new generic misoprostol (co-packaged with mifepristone) product will be bioequivalent to the original formulation misoprostol. 3.9 References Ashok PW, Templeton A, Waagarachchi PT, Flett GM. (2002) Factors affecting the outcome of early medical abortion: a review of 4132 consecutive cases. Brit J Obstet Gynaecol 109:1281-1289
El-Refaey H, Rajasekar D, Abdalla M, Calder L, Templeton A. (1995) Induction of abortion with mifepristone (RU486) and oral or vaginal misoprostol. New Eng J Med 332:983-987 European Medicines Agency (2002). Note for guidance on the investigation of bioavailability and bioequivalence. http://www.egagenerics.com/doc/emea_bioequiv-1401-98.pdf
Hamoda H, Ashok PW, Dow J, Flett GM, Templeton A. (2003) A pilot study of mifepristone in combination with sublingual or vaginal misoprostol for medical termination of pregnancy up to 63 days gestation. Contraception, 68:335-8 Hamoda H, Ashok PW, Flett GM, Templeton A. (2005) A randomised controlled trial of mifepristone in combination with misoprostol administered sublingually or vaginally for medical abortion up to 13 weeks of gestation. Brit J Obstet Gynaecol, 112:1102-8
RCOG, Royal College of Obstetricians and Gynaecologists. (2004) The care of women requesting induced abortion: Evidence-based guideline No7. London, RCOG Press Tang OS, Schweer H, Seyberth NW, Lee SWH, Ho PC. (2002) Pharmacokinetics of different routes of administration of misoprostol. Human Reprod 17:332-6
Tang OS, Chan CC, Ng EH, Lee SW, Ho PC. (2003) A prospective, randomized, placebo-controlled trial on the use of mifepristone with sublingual or vaginal misoprostol for medical abortions of less than 9 weeks gestation. Human Reprod, 18:2315-2318. World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (1993). Termination of pregnancy with reduced doses of mifepristone (RU486) in late first trimester abortion. Contraception 50:461-475 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2000). Comparison of two doses of mifepristone in combination misoprostol for early medical abortion; a randomized trial. Brit J Obstet Gynaecol 107:524-530 World Health Organization Task Force on Post-ovulatory Methods for Fertility Regulation (2003) WHO multinational study of three misoprostol regimens after mifepristone for early medical abortion. I: Efficacy. Brit J Obstet Gynaecol 110:808-818
Yu Zou, Xiaoyan Chen, Bo Song, Dafang Zhong (2007). Determination of misoprostol acid in human plasma by liquid chromatography coupled to tandem mass spectrometry. J Chromatog B, in press (Available online 13 January 2007)
Note: Start on this page and continue additional sheets as necessary.
Approvals will be obtained from the Ethics Committee of Gynaecology and Obstetrics, Otology, Ophtalmology, Neurology and Neurosurgery of the Hospital District of Helsinki and Uusimaa; and from the Finnish National Agency for Medicines. The study will be performed according to the Declaration of Helsinki.
All potential volunteers will be informed about the aims and protocol of the study, the side-effects that may occur as a result of the treatment. The measures taken to ensure confidentiality and their right to withdraw from the study at any time without prejudice to their further medical care will also be explained to them. Women participating in the study will be asked to sign the consent form.
Experience gained so far with the use of mifepristone followed by misoprostol for the termination of early pregnancy indicates that the therapy has side effects and a frequency of short-term complications comparable to those observed after vacuum aspiration. The most common complaint during treatment is lower abdominal pain, which results from uterine contractions and the abortion process itself – this is felt by practically all women. Using misoprostol, pregnancy-related symptoms, such as breast tenderness, nausea and vomiting decrease significantly during treatment and some women experience diarrhoea or shivering/fever during treatment. Less frequent side effects include dizziness, headache and rash. Side effects usually occur within the first two hours after misoprostol and are of low intensity, except for pain, which requires treatment in about half the cases.
Subjects will be informed about possible complications and qualified personnel and facilities will be available at all times to deal with them.
Most women participating in the study can be expected to have a complete abortion and
therefore will not be exposed to risks associated with vacuum aspiration, particularly the risks of physical trauma (cervical laceration, uterine perforation, etc). Even if surgical intervention is required in a small number of subjects, it is likely to be easier since the treatment will have dilated and softened the cervical canal.
No financial incentives will be offered to potential study participants and no financial remuneration will be given to women recruited into the study, other than reimbursement of expenses incurred as a result of participating in the study (travel expenses for extra hospital visits; lost days of work; and outpatient clinic fees). The subjects will be provided with a clinical follow-up, 2-3 weeks after the abortion.
Note: Ensure all information requested in the corresponding section of Part 1 is provided