Kamagra enthält Sildenafilcitrat als pharmakologisch aktiven Bestandteil. Dieser hemmt selektiv die Phosphodiesterase-5 und erhöht dadurch die Konzentration von cGMP im Corpus cavernosum. Der Effekt ist zeitlich begrenzt, da die Halbwertszeit von Sildenafil etwa vier Stunden beträgt. In der galenischen Form als Mundgel erfolgt die Resorption besonders rasch, was zu einem schnelleren Wirkeintritt führt. Der Abbau erfolgt überwiegend hepatisch über CYP3A4, wobei ein aktiver Metabolit entsteht, der zur Gesamtwirkung beiträgt. Typische Nebenwirkungen ergeben sich aus der Vasodilatation, darunter leichte Kopfschmerzen und nasale Kongestion. In klinischen Beschreibungen wird kamagra oral jelly im Zusammenhang mit der schnelleren Absorption erwähnt.
No job name
Comparison of 2D Similarity and 3D Superposition. Application to Searching a Conformational Drug Database
Martin Thimm,*,† Andrean Goede,‡ Stefan Hougardy,† and Robert Preissner‡
Institut fu¨r Informatik, Humboldt-Universita¨t zu Berlin, 10099 Berlin, Germany, and Institut fu¨r Biochemie,
Charite´, Monbijoustrasse 2, 10117 Berlin, Germany
In a database of about 2000 approved drugs, represented by 105 structural conformers, we have performed2D comparisons (Tanimoto coefficients) and 3D superpositions. For one class of drugs the correlation betweenstructural resemblance and similar action was analyzed in detail. In general Tanimoto coefficients and 3Dscores give similar results, but we find that 2D similarity measures neglect important structural/funtionalfeatures. Examples for both over- and underestimation of similarity by 2D metrics are discussed. The requiredadditional effort for 3D superpositions is assessed by implementation of a fast algorithm with a processingtime below 0.01 s and a more sophisticated approach (0.5 s per superposition). According to the improvementof similarity detection compared to 2D screening and the pleasant rapidity on a desktop PC, full-atom 3Dsuperposition will be an upcoming method of choice for library prioritization or similarity screeningapproaches.
based procedures explore configuration data from crystal-lographic databases;18 others emphasize the distinct features
The accessibility of large compound databases has changed
of bound ligand conformations especially for the muscarinic
from exclusive inhouse databases of large pharmaceutical
acetyl-choline receptor.19 Simulated annealing for difference
companies to inexpensive publicly available sources.1 At this
minimization20 or clustering procedures21 for better coverage
time about two million different compounds can be purchasedfrom different vendors.2 In this context established methods
of the low-energy conformational space22 are applied.
such as 2D similarity searching are increasingly applied to
The selection of the right features for the prediction of
identify active compounds for experimental assays. It was
bioactivity requires compound class specific techniques to
generally accepted that similar compounds having Tanimoto
obtain reasonable performance.23 It was shown that the
coefficients larger than 0.85 will exhibit similar biological
inclusion of 3D information via 3D field descriptors generates
activity.3 This assumption could be reaffirmed at a lower
level of 80% similar activity.4 In different assays the fraction
Typical 3D QSAR studies in this field are restricted to a
of active 0.85 similars declined to 60%-40%.5 In a recent
limited set of compounds matching the pharmacophore
analysis considering more than a hundred different assays
model.25,26 To complement this technique shape-based ap-
the estimation of the chance that a compound that is > 0.85
proaches are implemented27 and successfully applied to
Tanimoto similar to an active is itself active was further
similar problems as considered in the analysis of this paper.28
reduced to 30%.6 The resulting risk of missing attractivecompounds gives rise to a number of analyses comparing
The similarity is in the eye of the beholder, as Kubinyi
different molecular descriptors and similarity metrics for
illustrated.29,30 The scoring of the 3D similarity remains
different purposes.7,8 But nevertheless even the accuracy of
difficult because the balance between geometrical and
the prediction of one of six drug classes remains at 66%.9
physicochemical31 terms may influence the results toward
Descriptors representing 3D information10,11 and pharma-
scaffold hoppers32 or R-group similarities.33 For this analysis
cophore based approaches12,13 are opportunities to overcome
we selected the drug class of the neuroleptics because these
the weaknesses of 2D descriptors. However a lot of experi-
compounds are known to have a number of potential side
ence and intuition has to be invested to achieve reasonable
effects such as extrapyrimidal adverse events. The therapeutic
results.14 The superposition of 3D structures is a time-
action of neuroleptics is mediated by their interaction with
consuming task. For an extensive review on methods for
transmitter receptors in particular with the subtypes of the
structural alignment see ref 15 and the references therein.
dopamine-receptor. Here we focus on side effects that can
Structural flexibility has to be taken into account. The latter
be explained by the affinity to further receptors: histamine-,
problem can be approached either during comparison16 or
serotonin-, adrenergic, and muscarinic receptor.34 This can
prior to comparison.17 Different approaches for the generation
roughly be estimated by the similarity with compounds from
of conformers exhibit strengths and weaknesses. Knowledge
indication classes directly addressing these receptors suchas antipsychotics, psychoanaleptics, or antihistamines. The
* Corresponding author e-mail: thimm@informatik.hu-berlin.de. †
drug classification scheme according to the WHO recom-
mendation35 was utilized to this end.
Published on Web 00/00/0000 PAGE EST: 6.9
Table 1. Antipsychotics Used for the Data Base Search: Name,
• the correlation between 2D and 3D similarity,
• the fitness of Tanimoto coefficients for the drug class
• whether a simple geometric score will be useful as 3D
• whether 3D superposition will be useful to detect similar
Drug Classification. Recently, the recommendations of
the WHO Expert Committee responsible for updating theWHO Model List of Essential Medicines were published.35For the first time, a list of all items on the Model List sortedaccording to their 5-level Anatomical Therapeutic Chemical(ATC) classification codes was given. As the therapeuticsubgroup is determined by the second level and the chemicalcomponent describes the lower level(s) of classification it isuseful for this type of analysis, correlating structural similar-ity with similar therapeutic action.
The pharmacological action of neuroleptics is mediated
by their interaction with transmitter receptors in particularwith the subtypes of the dopamine-receptor. Therefore(particular) neuroleptics are known to have a number of sideeffects and are dubbed “dirty drugs”.36 Here we focus oneffects that can be explained by the affinity to furtherreceptors: histamine-receptor (H1), serotonin-receptors (5-HT2A/B,5-HT3), adrenergic receptor (alpha 1), and musca-rinic acetyl-choline receptor (M).34 This can roughly beestimated by the similarity with compounds from indicationclasses directly addressing these receptors: N05A (antipsy-chotics), N06 (psychoanaleptics), D04/R06 (antihistamines,
• paths of different lengths (2 to 7) between atoms of the
same type and same order of the bonds, e.g. CdCN, CCd
Data. All comparisons are performed on a database of
N for a path of length 2, or OdCCdN for a path of length
2086 3D-structures of drugs extracted from our inhouse
database. This complies with the number of approved drugs
Because of the limited length of the fingerprint it is not
included in the ChemIDplus database,37 which contains a total
possible to assign a special bit for only one pattern. Instead
of 177 000 chemical structures. To improve the conditions
of this each pattern is assigned a small number of positions
for the 3D-comparisons 85 800 conformers were computed
(say 4 or 5) along the fingerprint which are set to 1. Therefore
with Catalyst38 according to the algorithm of Smellie.39 The
the fingerprints of two molecules can be the same while the
Anatomical Therapeutic Chemical (ATC) Classification
molecules are different. Additionally, the positions corre-
System is used for the classification of drugs. It is controlled
sponding to a special pattern account for the occurrence of
by the WHO Collaborating Centre for Drug Statistics
the pattern. Multiple appearances of the same pattern give
Methodology40 and was first published in 1976. The database
the same fingerprint. Therefore featureless molecules (such
covers 218 ATC-major classes (like N05A). 185 ATC-major
as C20H22 or C30H32) give the same fingerprint. Neverthe-
classes are represented by at least 3 structures, this meets
less the fingerprints indicate whether a compound can be a
98% of all such classes containing at least 3 different actual
substructure of another molecule. The Tanimoto coefficient
low molecular weight compoundsswithout e.g. combina-
between two fingerprints is the proportion of the bits in
tions, bandages, or proteins. Table 1 shows the 13 members
common and the bits in at least one fingerprint
of the ATC-class N05A (antipsychotics) that were used forthe database search. 2D-Comparison. The 2D-comparison of the molecules
was carried out using the Tanimoto coefficients41 computed
where BC is the number of bits which are 1 in both
by the corresponding procedure of Accord from Accelrys.38
fingerprints, whereas B1 and B2 are the number of bits which
For this reason the fingerprints of the structures are calculated
are 1 in the first or the second fingerprint, respectively.
using the Daylight algorithm42 and compared by the Tan-
3D Score. The 3D-superposition-algorithms investigated
imoto similarity measure for bit strings. Fingerprints are
here are designed to find spatial similarity between mol-
Boolean arrays of a given length. To evaluate the fingerprint
ecules. We follow the paradigm that a necessary condition
each pattern of the molecule is generated. Such patterns are
for functional similarity is similar geometry. For this reason
the scoring function is built to measure spatial similarity only.
• bonds (single, double,.) between atoms of special types
By this it is possible to find superpositions which we would
COMPARISON OF 2D SIMILARITY AND 3D SUPERPOSITION
not have found by simultaneously trying to incorporate
One good news is that the second subproblem is known
physicochemical features. So it may happen that few of the
to be solvable in polynomial time.43 A way to solve the
geometrically found hits turn out to be biologically irrelevant,
superposition problem is to enumerate all possible assigments
but we considerably lower the risk of losing most interesting
and to compute the rigid motion for each of them. But here
and relevant hits that come from different chemical structures.
the bad news is that the number of possible assignments
Similarity of molecules is measured by superposition. In
grows highly exponential in the number of atoms. With this
general the superposition problem may be decomposed into
naive approach only instances with very few atoms (less than
two subproblems: First, we have to find an assignment (or
matching) of the atoms of one molecule to the atoms of the
With the help of a branch-and-bound approach, a widely
other molecule, that tells us which atom on one side has to
used technique in optimization, we are able the reduce the
be superimposed with which atom on the other side (or not
number of assignments to be tested dramatically. Doing this
superimposed at all). Second, a rigid motion is computed to
we are able to solve the superposition problem up to
optimally perform this action. There are two competing
optimality quite fast (10 atoms: few seconds, 16 atoms: 1-2
1. The more atoms are actually superimposed the better
Since drug-like molecules are often larger and since the
above-mentioned running times are still far too slow we have
2. The distances of the matched atoms should be as small
to find ways to overcome these difficulties. We can no longer
as possible. A way to balance these two goals is the following
hope to solve the problem exactly, i.e., the solution of the
scoring function: Consider two molecules A and B with m
algorithm described next will not be guaranteed to be the
and n atoms, respectively (m e n). Given an assignment M
best possible, but we will get it very quickly without losing
that maps the atoms aM of A, i ) 1,., k, k
much quality. In what follows we describe the two algorithms
atoms bM, j ) 1,., k. The resulting superposition has the
Fast 3D-Superposition. The algorithm presented here can
roughly be sketchted by the following steps:
score(A, B, M) ) k e(-rmsd(M))
2. orientation according to principal moments of inertia
The first term, k/m, measures the proportion of actually
The first orientation (in step 2) is of course independent
superimposed atoms of the smaller molecule, the second
of transformations of the coordinate system and quite stable
term, the root-mean-square distance of these atoms
for small alterations of the atomic positions. The normaliza-tion of the atomic sets is unique except for possible rotations
(original arrangement and rotations of 180° around the x-,
y-, or z-axis). This means that the degree of freedom is
strongly reduced and the assignment of pairs of atoms isrelatively straightforward for identical and slightly modified
quantifies the distance of matched atoms.
atomic sets because only four possible normalizations have
By this definition we try to find a superposition with many
to be checked to identify related atoms: imagine determining
atoms superimposed with small distance. The first term
the correct orientation of a credit card (magnetic strip: top
increases with the number of superimposed atoms, but in
surface, right; top surface, left; bottom surface right; bottom
the majority of cases this will make the second term decrease
surface, left). In a first step the centers of mass of the two
since more assigned atoms will result in higher rmsd.
atomic sets are determined. All the coordinates of the atoms
Observe that the value of the scoring function is always
included are transformed to superimpose the centers of mass.
between 0 and 1, where larger values mean higher similarity.
To determine the least and largest (orthogonal) expansion,
The scoring function is not restricted to molecules of the
the plane and the straight line of minimal quadratic distance
same size. It is also possible to compare molecules of quite
to all atoms have to be computed. The normal line of the
different size, since the first term of our scoring function
plane gives the least expansion and the straight line of
allows us to find smaller molecules inside larger ones.
minimal quadratic distance points at the largest expansion. 3D Comparison. Since no polynomial time algorithm is
Using these directions one atomic set is rotated such that
known to solve the superposition problem as described above
the major directions coincide. There are four possible
to optimality, we have to use heuristic methods to get good
normalizations for an atomic set that coincide with the
solutions in reasonable time. We have implemented two
exception of 180° rotations around the x-, y-, or z-axes. In a
different approaches, a fast one and a more sophisticated
further step all four normalizations are used to determine
the pairs of atoms between the two atomic sets. This
In general the superposition problem may be decomposed
normalization procedure is stable even if additional atoms
into two subproblems: First, find the assignment for the two
are included in one of the sets. Therefore, the normalization
atom sets, i.e., which atom in one molecule should be
of the atomic set can be used to identify pairs of correspond-
superimposed with which atom in the other molecule.
ing atoms. Two atoms form a pair if they are mutually the
Second, find the rigid motion that gives the optimal
nearest atoms, and their distance is lower than a given cutoff
superposition of the atom sets, given the assignment. Our
value. Different cutoff values were tested showing that a
two approaches mainly differ in the effort made to solve the
cutoff of 2.5 Å performed best for sets of densely packed
atoms. For all four normalizations the number of atom pairs
is chosen, and the root-mean-square distance (rmsd) is
• step 2 Enumerate all possible assignments of these s
calculated for the related atomic pairs. The normalizations
atoms to their nearest neighbors (including the possibility
are weighted on the basis of these values. The best
that an atom is assigned to have no matching partner), carry
normalization (largest number of pairs) is used in a further
out the appropriate rigid motion, and pick the one with the
step to improve the alignment. For the given set of pairs the
best score value (on this partial instance of already fixed
optimal superposition is estimated, followed by a new search
of related pairs until the assignment of the atoms does not
• step 3 Fix the assignment on these s atoms and perform
the implied rigid motion to all atoms. Go to step 1 while
Sophisticated 3D-Superposition. The second algorithm
there are atoms that are not yet fixed.
We conclude this section with some implementation
1. Reduce the given instance to a smaller artificial instance
details. Since the solution of Phase 1, the artificial pseudo-
that is in a certain sense similar to the original one.
molecule, may look quite different for different numbers r,
2. Solve this artificial instance optimally with the above-
we perform this step for several different values of r. The
mentioned branch-and-bound technique.
optimal solution of Phase 2 may not be exactly what we
3. Lift the solution of the smaller, artificial instance to a
want, since we observed in numerous tests of the exact
solution for the original problem. Next we describe the three
algorithm that there are instances that have quite a number
of solutions with very similar score values but very different
Phase 1: Reduction to a smaller artificial instance
assignments. To overcome this we store not only the best
The running time of the exact algorithm mainly depends
solution but the best n of them (seen during the branch-and-
on the number of atoms of the two molecules; so the aim of
bound process). Phase 3 also depends on the predefined
this phase is to construct, starting with the original molecules,
number s, so again as in Phase 1 we perform this phase for
new, artificial pseudomolecules with fewer pseudoatoms, that
several different values of s. As one would expect, the quality
are still spatially similar to the original molecules. This is
of the solution increases with the size of the r- and s-intervals
done iteratively in the following way, sometimes called
and with n, but so does the running time. (Running time
grows nearly proportional with n and the number of different
• Start with the original molecule, call every atom a
s-values and superproportional in the number of different
r-values, since larger r-values get more and more expensive.)
• While the number of pseudoatoms is larger than a
Our standard parameters for drug-like molecules are
r∈{3, 4, 5}, s∈{3, 4}, n ) 40. We determined them as a
- look for those two pseudoatoms with the smallest
result of numerous tests trying to find an optimal tradeoff
distance and merge them to a new pseudoatom. The
coordinates of this new pseudoatom are given by the
Although we cannot prove the optimality for our algo-
weighted center of gravity of the two merged ones, which
rithms, we wanted to see how far away we are in the relevant
cases. As a test we computed 3D-scores for quite a number
So in every such step the number of pseudoatoms is
of instances which were known to have large 3D-scores
decreased by one. As a remark we should say that we take
(>0.70) up to optimality with the branch-and-bound algo-
into account how many original atoms are represented by a
rithm mentioned above. (As a remark we should say that
pseudoatom by attaching weights to it. The idea is that the
this is possible in these casesswith still very large running
new instance constructed in this way still carries the spatial
timesssince branch-and-bound algorithms tend to find very
information of the original molecule to a certain extent.
good resultssif they existsquite “fast”.) The results showed
Phase 2: Exact solution of artificial instance
that in all cases the sophisticated approach was within five
Now we solve our artifical instance with the exact
algorithm. The solution of this step is the starting point ofPhase 3.
Phase 3: Lift of intermediate solution to the original instance
To compare the two 3D-superposition algorithms with the
The rigid motion which led to the solution in Phase 2 may
2D-approach we selected 13 antipsychotics (ATC code
also be applied to the original instance. The idea of our
N05A) and computed both Tanimoto coefficients and 3D-
approach is now that, since the smaller artificial instance is
scores (fast and sophisticated) for each of them with all drugs
spatially similar to the original one, the position of the
in the database (with more than 14 non-hydrogen atoms).
original atoms after this rigid motion is not far from a very
To compute the 3D-score of two conformers of molecules
good solution. We only have to refine the assignment to these
of average size (25 atoms) we need about 0.01 s (fast) and
atoms. This is done as follows: The distance of atoms that
0.5 s (sophisticated) (on a 2GHz PC), resulting in a total
will be assigned to each other should now be already quite
running time of 25 s (fast) and 21 min (sophisticated) to
small, so a natural approach is the following:
fully compare two molecules, both given by 50 structural
• step 0 Declare all atoms to be not fixed.
• step 1 Sort those atoms of the first molecule that have
Unfortunately we cannot compare the values of Tanimoto
not yet been assigned (fixed) increasingly by their distance
coefficients and 3D-scores one-to-one. As mentioned in the
to the nearest neighbor in the other molecule, that is still
Introduction it is generally accepted that Tanimoto coef-
available. Take the first s of them (s is a small predefined
ficients larger than 0.85 start to indicate similar activity. To
find a corresponding value for the 3D-score we counted the
COMPARISON OF 2D SIMILARITY AND 3D SUPERPOSITION
Table 2. Number of Hits Table 3. Number of Hits: Tanimoto vs Soph. 3D Table 4. Number of Hits: Tanimoto vs Fast 3D Figure 1. Comparisons with 2D similarity above threshold and
3D similarity below threshold. (a) Superposition of flupentixol(bottom, ATC code: N05AF01) with thiethylperazine (top, ATCcode: R06AD03)
Table 5. Number of Hits: Fast 3D vs Soph. 3D
coefficient of 0.89. The similarity is underestimated by the 3D score
because the distortions in the tricyclic ring are not properlyrepresented by the conformers. (b) Superposition of tiapride (top,
ATC code: N05AL03) and probenecid (bottom, ATC code:
M04AB01) with a 3D score of 0.58 and a Tanimoto coefficient of
0.85. The resemblance to probenecid, an antigout drug, is oversti-
mated by the Tanimoto coefficient because of the identical chemicalsubgroups (phenyl, sulfonyl).
number of hits with a Tanimoto coefficient larger than 0.85and found that a 3D-score of about 0.75 gives approximately
hits, that are clearly relevant from a biological point of view,
we can infer that already 3D-score above 0.65 are worth
We found 164 hits with a Tanimoto coefficient larger than
while to look at. (See Figure 1a) for an example.)
0.85. The 3D-superposition algorithms returned 200 (sophis-
The second subset of hits (i.e. 4. column) with large
ticated) and 156 (fast) hits with a 3D-score larger than 0.75
Tanimoto coefficents and small 3D-scores largely consists
of those hits for which the Tanimoto coefficient highly
Since we want to show that the 3D-approach is appropriate
overestimates the structural/functional similarity of the
to find molecules that have similar activity we first looked
molecules. An example can be seen in Figure 1b.
at the ATC codes of the hits with a score value larger than
Ad b. For this set of hits the situation changes. The number
0.75 and found that 78, 5% (157 out of 200) (sophisticated)
of hits with a large 3D-score that are found by the
and 84% (131 out of 156) (fast) of these can be found in
sophisticated algorithm are significantly larger than those
drug subclasses that are known to have similar activity or
found by the fast algorithm, for both the relevant drug classes
similar adverse reaction (ATC codes N05A, N06, R06, D04).
and the others (see Tables 3 and 4 for the exact numbers).
The proportion of hits in these classes for Tanimoto
Since the 3D-superposition algorithms are not designed
coefficients larger than 0.85 is 69% (113 out of 164) (see
to incorporate chemical features, there are some hits that are
clearly geometrically relevant, but perhaps their prediction
To compare the results of the 2D- and 3D-approaches we
about similar activity is quite limited. These hits can be found
have to look at three different sets of seemingly similar pairs
in the second subset (i.e. 4. column).
What we are really aiming for is the first subset (i.e. 3.
a. large Tanimoto coefficient (>0.85), small 3D-score
column). Here we find hits that are both geometrically similar
and relevant concerning prediction of similar activity and
b. small Tanimoto coefficient (<0.85), large 3D-score
function. One reason why in these cases Tanimoto coef-
ficients do not indicate similarity are slight changes in
c. large Tanimoto coefficient (>0.85), large 3D-score
chemical structure. Furthermore there are hits with two
molecules of somewhat different size. It is known (see ref
Ad a. For this set of hits, comparing Tanimoto coefficients
44) that for these instances Tanimoto coefficients are more
to both sophisticated and fast 3D-superposition gives a
and more inefficient, while our 3D-score is designed to also
similar picture (see Tables 3 and 4 for the exact numbers).
find these hits. (See some examples for both cases in Figure
Approximately one-half of this set of hits lies in the above-
2.) For this type of hits the sophisticated approach is clearly
mentioned relevant drug classes. A closer inspection for this
superior to the fast algorithm. Comparing the numbers in
subset (i.e. 3. column) shows that in most of the cases the
Table 5 with those in Tables 3 and 4 shows that most of the
3D-score for these hits is larger than 0.65. Looking at these
hits for which the two 3D-approaches differ can be found in
Figure 2. Comparisons with low Tanimoto coefficients and 3D Figure 3. Differences between fast and sophisticated superposi-
scores above threshold. (a) Superposition of fluphenazine (bottom,
tions. (a) Superposition of perazine (top, ATC code: N05AD10)
ATC code: N05AB02) with isothipendyl (top, ATC codes: D04A-
with triflupromazine (bottom, ATC code: N05AA05) with a 3D
A22, R06AD09) with a 3D score of 0.81 and a Tanimoto coefficient
score of 0.77 (sophisticated), 0.50 (fast), and a Tanimoto coefficient
of 0.69. The resemblance to isothipendyl, an antihistaminic agent,
of 0.84. The resemblance between the two neuroleptics is neglected
is neglected by the 2D similarity measure because of missing
by the fast superposition algorithm because the centers of gravity
chemical groups (trifluoromethyl, piperazin) and quite different sizes
do not fit. (b) Superposition of tryptophan (top, ATC code:
of the molecules. (b) Superposition of prothipendyl (bottom, ATC
N06AX02) and risperidone (bottom, ATC code: N05AX08) with
code: N05AX07) and opipramol (top, ATC code: N06AA05) with
a 3D score of 0.77 (sophisticated), 0.50 (fast), and a Tanimoto
a 3D score of 0.76 and a Tanimoto coefficient of 0.72. The
coefficient of 0.44. The similarity to tryptophan, an antidepressant,
similarity to opipramol, an antidepressant, is missed by 2D
is missed by the fast superposition algorithm because of the very
comparison because the middle ring is seven membered in
different overall geometry of the molecules.
opipramol (dibenzazepine derivative) and six membered in pro-thipendyl (azaphenothiazine derivative).
cal features that are responsible for the biological activity.
the class discussed here. The main reason for this is that the
2D similarity works poorly when common functional groups
fast approach is not able to find hits for two molecules that
as in peptides are considered. A similar fragment- or
have different overall geometry, in particular small molecules
topomer-based steric shape screening was shown to be more
that are substructures of larger ones are not found (see Figure
selective than 2D similarity,13 especially advantageous “lead-
hopping” was observed. A reasonable speed for the in silico
Ad c. In this set we find those hits that are quite similar
screening of large compound libraries can be achieved by
in both chemical structure and size. They are reported as
full-atom superposition procedures as presented in this
relevant by both approaches. For this type of hits both
strategies are most similar. Here again, as in case a, the fast
With receptor structures available ligand-docking programs
and the sophisticated 3D-algorithm perform comparably.
have been shown to enrich hit lists of in silico screening
From our point of view we have therefore seen several
approaches,45 but in the case of psycholeptics a number of
strong arguments in favor of the 3D-superposition algorithms.
structurally unknown receptors are engaged. Most of the
It can be clearly seen that the 3D-approach is able to detect
processes involved in ADME are driven by rather unspecific
similar activity and similar adverse reaction, even with this
interactions between drugs and macromolecules, but drug
seemingly simple, purely geometry-based scoring function.
transporters and cytochromes gained increased interest in
For large data sets a fast 3D-superposition algorithm
early ADME profiling via similarity based structure activity
combined with Tanimoto coefficients helps to increase the
relation (SIBAR).46 The increased predictive power of the
3D- vs 2D-similarity for side effects demonstrated in this
If one aims to really find all, at least geometrically relevant
analysis gives rise to the hope that improvements in ADME
hitssthis may be important for smaller and more specific
and toxicity profiling will be possible.
sets of moleculessit is worthwhile to follow the sophisticated
Limitations of the fast 3D superposition approach are
3D-approach (with a somewhat smaller threshold for rel-
spherical compounds for which it might fail to find proper
evance). We were able to find really relevant hits that cannot
assignments. The known size bias and size limitation of 2D
be found by simple 2D-methods or by the fast 3D-algorithm.
similarity measures44 also may cause problems for the fastalgorithm.
The conformer generation is a general problem because
In agreement with our results it is shown in refs 27 and
the 3D similarity between two structural ensembles depends
28 that 3D similarity searches retrieve compounds with more
critically on the original structures, the conformer genera-
diverse topology, while 2D similarity works best when the
tion22 and clustering47 algorithm, the parameters such as
query molecule contains relatively rare and distinct topologi-
energy threshold, and the number of conformers per com-
COMPARISON OF 2D SIMILARITY AND 3D SUPERPOSITION
pound. In particular the number of rotatable bonds will
(21) Barnum, D.; Greene, J.; Smellie, A.; Sprague, P. Identification of
restrict the 3D similarity approach or will require new
common functional configurations among molecules. J. Chem. Inf. Comput. Sci. 1996, 36, 563-571.
(22) Smellie, A.; Stanton, R.; Henne, R.; Teig, S. Conformational analysis
by intersection: CONAN. J. Comput. Chem. 2003, 24, 10-20.
(23) Weston, J.; Perez-Cruz, F.; Bousquet, O.; Chapelle, O.; Elisseeff, A.;
Scholkopf, B. Feature selection and transduction for prediction of molecular bioactivity for drug design. Bioinformatics 2003, 19, 764- 771.
Stefan Hougardy and Martin Thimm are supported by the
(24) Schuffenhauer, A.; Gillet, V. J.; Willett, P. Similarity searching in
files of three-dimensional chemical structures: analysis of the BIO-
DFG Research Center “Mathematics for key technologies”,
STER database using two-dimensional fingerprints and molecular field
and Andrean Goede and Robert Preissner are supported by
descriptors. J. Chem. Inf. Comput. Sci. 2000, 40, 295-307.
(25) Bostrom, J.; Bohm, M.; Gundertofte, K.; Klebe, G. A 3D QSAR study
the BMBF funded Berlin Center of Genome Based Bioin-
on a set of dopamine D4 receptor antagonists. J. Chem. Inf. Comput.Sci. 2003, 43, 1020-1027.
(26) Bostrom, J.; Gundertofte, K.; Liljeforsa, T. A pharmacophore model
for dopamine D4 receptor antagonists. J. Comput.-Aided Mol. Des.2000, 14, 769-786.
(27) Hahn, M. Three-Dimensional Shape-Based Searching of Conforma-
(1) Voigt, J. H.; Bienfait, B.; Wang, S.; Nicklaus, M. C. Comparison of
tionally Flexible. J. Chem. Inf. Comput. Sci. 1997, 37, 80-86.
the NCI open database with seven large chemical structural databases.
(28) Guner, O. F.; Hahn, M.; Li, H.; Hassan, M. 2D versus 3D shape
J. Chem. Inf. Comput. Sci. 2001, 41, 702-712.
similarity: use of molecular shape-based 3D searching techniques for
(2) Bradley, M. P. An overview of the diversity represented in commeri-
identifying novel compounds. Case study, Accelrys; http://www.
cally-available databases. Mol. DiVers. 2002, 5, 175-183.
accelrys.com/cases/2dvs3di_full.html.
(3) Matter, H. Selecting optimally diverse compounds from structure
(29) Kubinyi, H. Molecular similarity. 1. Chemical structure and biological
databases: a validation study of two-dimensional and three-dimen-
action. Pharm. Unserer Zeit 1998, 27, 92-106.
sional molecular descriptors. J. Med. Chem. 1997, 40, 1219-1229.
(30) Kubinyi, H. Molecular similarity. 2. The structural basis of drug design.
(4) Brown, R. D.; Martin, Y. C. An evaluation of structural descriptors
Pharm. Unserer Zeit 1998, 27, 158-172.
and clustering methods for use in diversity selection. SAR QSAR
(31) Iwase, K.; Hirono, S. Estimation of active conformations of drugs by
EnViron. Res. 1998, 8, 23-39.
a new molecular superposing procedure. J. Comput.-Aided Mol. Des.
(5) Delaney, J. S. Assessing the ability of chemical similarity measures
1999, 13, 499-512.
to discriminate between active and inactive compounds. Mol. DiVers.
(32) Schneider, G.; Neidhart, W.; Giller, T.; Schmid, G. “Scaffold-Hopping”
1996, 1, 217-222.
by Topological Pharmacophore Search: A Contribution to Virtual
(6) Martin, Y. C.; Kofron, J. L.; Traphagen, L. M. Do structurally similar
Screening. Angew. Chem. Int. Ed. Engl. 1999, 38, 2894-2896.
molecules have similar biological activity? J. Med. Chem. 2002, 45,
(33) Holliday, J. D.; Jelfs, S. P.; Willett, P.; Gedeck, P. Calculation of
intersubstituent similarity using R-group descriptors. J. Chem. Inf.
(7) Chen, X.; Reynolds, C. H. Performance of similarity measures in 2D
Comput. Sci. 2003, 43, 406-411.
fragment-based similarity searching: comparison of structural descrip-
(34) Maurer, I.; Volz, H. P. Cell-mediated side effects of psychopharma-
tors and similarity coefficients. J. Chem. Inf. Comput. Sci. 2002, 42,
cological treatment. Arzneimittelforschung 2001, 51, 785-792.
(35) The selection and use of essential medicines. Report of the WHO
(8) Whittle, M.; Willett, P.; Klaffke, W.; van Noort, P. Evaluation of
Expert Committee, 2002 (including the 12th Model list of essential
similarity measures for searching the dictionary of natural products
medicines). World Health Organ. Tech. Rep. Ser. 2003, 914, i-vi,
database. J. Chem. Inf. Comput. Sci. 2003, 43, 449-457.
(9) Dixon, S. L.; Merz, K. M., Jr. One-dimensional molecular representa-
(36) Uhl, G. R.; Vandenbergh, D. J.; Miner, L. L. Knockout mice and dirty
tions and similarity calculations: methodology and validation. J. Med.
drugs. Drug addiction. Curr. Biol. 1996, 6, 935-936. Chem. 2001, 44, 3795-3809.
(37) Specialized Information Services (SIS) Division of the National Library
(10) Wildman, S. A.; Crippen, G. M. Three-dimensional molecular descrip-
of Medicine (NLM), 8600 Rockville Pike, Bethesda, http://chem-
tors and a novel QSAR method. J. Mol. Graphics Modell. 2002, 21,
(38) Accelrys Inc., San Diego, CA. http://www.accelrys.com.
(11) Turner, D. B.; Willett, P. The EVA spectral descriptor. Eur. J. Med.
(39) Smellie, A.; Stanton, R.; Henne, R.; Teig, S. Conformational analysis
Chem. 2000, 35, 367-375.
by intersection: CONAN. J. Comput. Chem. 2003 Jan 15; 24(1): 10-
(12) Hecker, E. A.; Duraiswami, C.; Andrea, T. A.; Diller, D. J. Use of
catalyst pharmacophore models for screening of large combinatorial
(40) WHO Collaborating Centre for Drug Statistics Methodology, http://
libraries. J. Chem. Inf. Comput. Sci. 2002, 42, 1204-1211.
(13) Cramer, R. D.; Jilek, R. J.; Andrews, K. M. Dbtop: topomer similarity
(41) Willett, P.; Barnard, J. M.; Downs, G. M. Chemical similarity
searching of conventional structure databases. J. Mol. Graphics Modell.
searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983-996. 2002, 20, 447-462.
(42) Daylight Chemical Information Systems, Santa Fe, NM. http://
(14) Patel, Y.; Gillet, V. J.; Bravi, G.; Leach, A. R. A comparison of the
pharmacophore identification programs: Catalyst, DISCO and GASP.
(43) Umeyama, S. Least-Squares Estimation of Transformation Parameters
J. Comput.-Aided Mol. Des. 2002, 16, 653-681.
Between Two Point Patterns. IEEE Trans. Pattern Anal. Machine
(15) Lemmen, C.; Lengauer, T. Computational methods for the structural
Intelligence 1991, 13, 676-681
alignment of molecules. J. Comput.-Aided Mol. Des. 2000, 14, 215-
(44) Holliday, J. D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display
of the size dependence of chemical similarity coefficients. J. Chem.
(16) Lemmen, C.; Lengauer, T.; Klebe, G. FLEXS: a method for fast
Inf. Comput. Sci. 2003, 43, 819-828.
flexible ligand superposition. J. Med. Chem. 1998, 41, 4502-4520.
(45) Jenkins, J. L.; Kao, R. Y.; Shapiro, R. Virtual screening to enrich hit
(17) Kramer, A.; Horn, H. W.; Rice, J. E. Fast 3D molecular superposition
lists from high-throughput screening: a case study on small-molecule
and similarity search in databases of flexible molecules. J. Comput.-
inhibitors of angiogenin. Proteins 2003, 50, 81-93. Aided Mol. Des. 2003, 17, 13-38.
(46) Klein, C.; Kaiser, D.; Kopp, S.; Chiba, P.; Ecker, G. F. Similarity
(18) Klebe, G.; Mietzner, T.; Weber, F. Methodological developments and
based SAR (SIBAR) as tool for early ADME profiling. J. Comput.-
strategies for a fast flexible superposition of drug-size molecules. J.Aided Mol. Des. 2002, 16, 785-793. Comput.-Aided Mol. Des. 1999, 13, 35-49.
(47) Raymond, J. W.; Blankley, C. J.; Willett, P. Comparison of chemical
(19) Furukawa, H.; Hamada, T.; Hayashi, M. K.; Haga, T.; Muto, Y.; Hirota,
clustering methods using graph- and fingerprint-based similarity
H.; Yokoyama, S.; Nagasawa, K.; Ishiguro, M. Conformation of
measures. J. Mol. Graphics Modell. 2003, 21, 421-433.
ligands bound to the muscarinic acetylcholine receptor. Mol. Phar-
(48) Raymond, J. W.; Willett, P. Similarity Searching in Databases of
macol. 2002, 62, 778-787.
Flexible 3D Structures Using Smoothed Bounded Distance Matrices.
(20) Mills, J. E.; de Esch, I. J.; Perkins, T. D.; Dean, P. M. SLATE: a
J. Chem. Inf. Comput. Sci. 2003, 43, 908-916.
method for the superposition of flexible ligands. J. Comput.-Aided Mol. Des. 2001, 15, 81-96.
Reference Guide for Pharmacy Technician Exam-Second Edition MANAN SHROFF Reference Guide for Pharmacy Technician Exam-Second EditionThis reference guide is not intended as a substitute for the advice of a physician. Students or readers mustconsult their physicians about any existing problem. Do not use any information in this reference guide forany kind of self treatment. Do not administer a
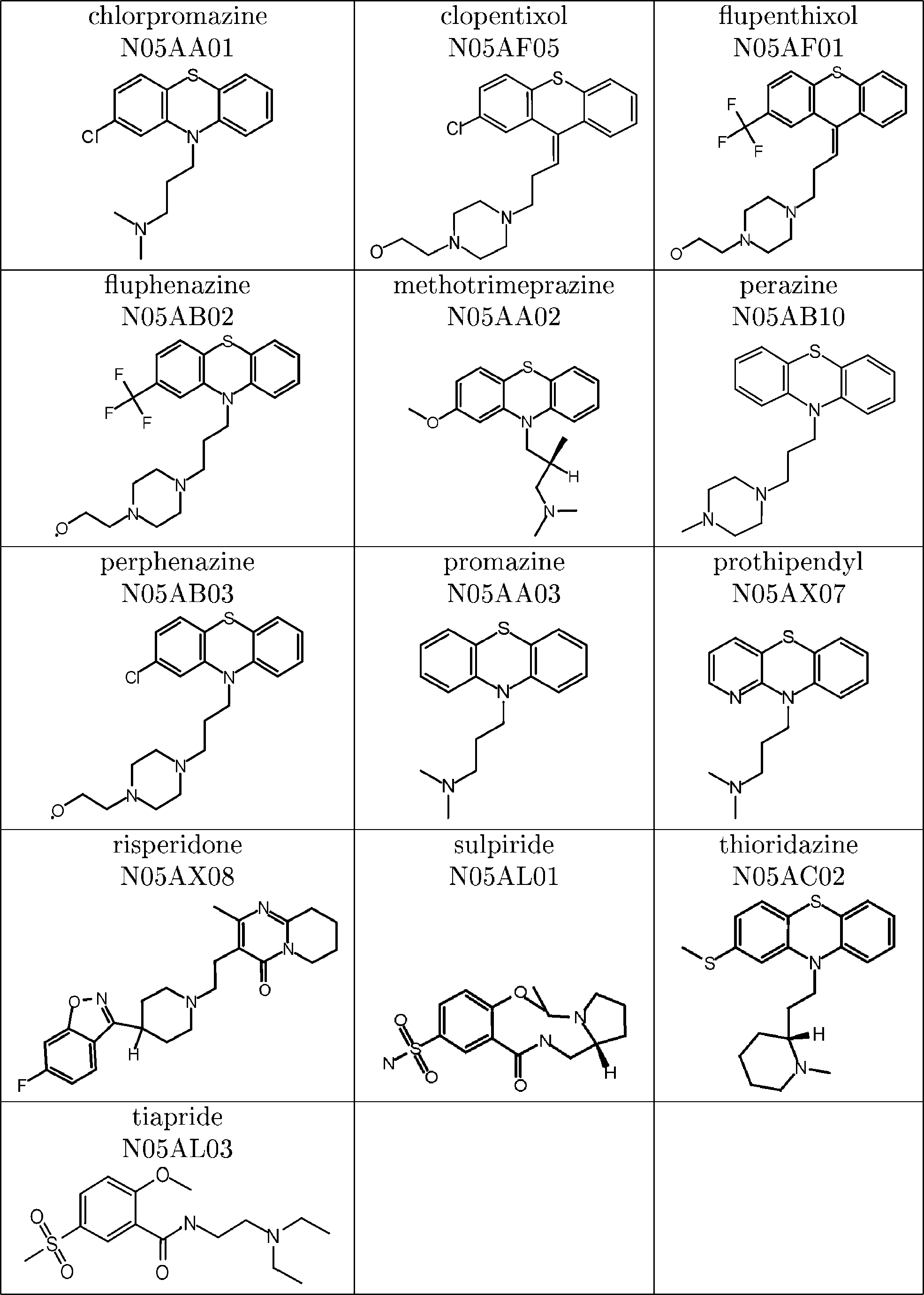 Table 1. Antipsychotics Used for the Data Base Search: Name,
Table 1. Antipsychotics Used for the Data Base Search: Name,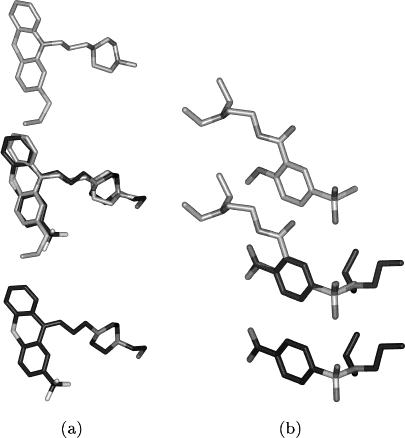 COMPARISON OF 2D SIMILARITY AND 3D SUPERPOSITION
Table 2. Number of Hits
COMPARISON OF 2D SIMILARITY AND 3D SUPERPOSITION
Table 2. Number of Hits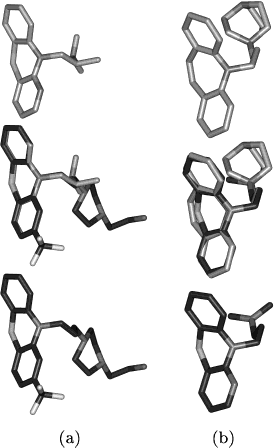
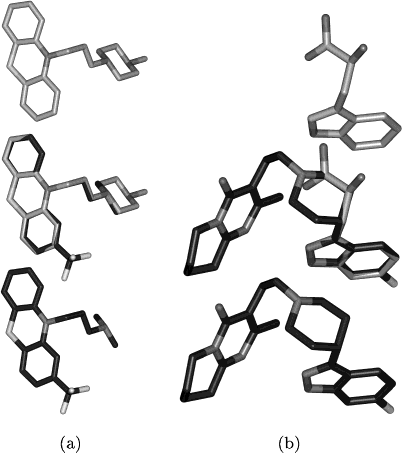 Figure 2. Comparisons with low Tanimoto coefficients and 3D
Figure 2. Comparisons with low Tanimoto coefficients and 3D