Kamagra enthält Sildenafilcitrat als pharmakologisch aktiven Bestandteil. Dieser hemmt selektiv die Phosphodiesterase-5 und erhöht dadurch die Konzentration von cGMP im Corpus cavernosum. Der Effekt ist zeitlich begrenzt, da die Halbwertszeit von Sildenafil etwa vier Stunden beträgt. In der galenischen Form als Mundgel erfolgt die Resorption besonders rasch, was zu einem schnelleren Wirkeintritt führt. Der Abbau erfolgt überwiegend hepatisch über CYP3A4, wobei ein aktiver Metabolit entsteht, der zur Gesamtwirkung beiträgt. Typische Nebenwirkungen ergeben sich aus der Vasodilatation, darunter leichte Kopfschmerzen und nasale Kongestion. In klinischen Beschreibungen wird kamagra oral jelly im Zusammenhang mit der schnelleren Absorption erwähnt.
Atrofia de werdnig hoffmann
Residente de tercer año de Pediatría, HIGA San José de Pergamino
Lactante de sexo masculino de dos meses de edad, que ingresa a nuestra sala de Pediatría con diagnóstico de atelectasia masiva. Refiere cuadro de 72 hs de evolución caracterizado por dificultad respiratoria e hiporexia.
Antecedentes personales: fruto de un embarazo controlado, G1 P1, parto eutócico, RNT de 40 semanas de gestación, peso: 3200 gr., score de Apgar vigoroso, internación conjunta. Caída tardía del cordón umbilical -a los 24 días de vida-. Lactancia materna exclusiva, con esquema de vacunación acorde a la edad.
Antecedentes familiares: madre, 19 años, sana; padre 26 años, sano. No refieren antecedentes familiares de relevancia.
A su ingreso, el paciente se encontraba alerta, en regular estado general, afebril, hemodinámicamente compensado, taquipneico, taquicárdico, conectado con el medio. Su auscultación evidenciaba regular entrada de aire bilateral, con disminución del murmullo vesicular en hemitórax izquierdo, mecánica ventilatoria exigida, con tiraje subcostal e intercostal y patrón tusígeno débil. Saturación oxígeno 92%, aire ambiente.
Se realizaron estudios de laboratorio de rutina, que no arrojaron resultados patológicos y radiografía de tórax, donde se evidenciaba una radioopacidad en lóbulo superior izquierdo, que tracciona el mediastino hacia su lado, compatible con atelectasia. Se tomaron hemocultivos.
Se indicaron medidas de sostén, oxigenoterapia, SNG, salbutamol 200mcg cada 4 hs, hidrocortisona 18mg/k, dexametasona 0,6mg./Kg./día, ampicilina 200 mg./Kg./día, gentamicina 5 mg./kg./día y kinesioterapia respiratoria.
Si bien el paciente ingresó por dificultad respiratoria, ciertos datos de su examen neurológico llamaban la atención, siendo francamente patológicos. En decúbito dorsal, el lactante se encontraba con sus cuatro extremidades en
extensión e hipotónicas; con tortícolis izquierda, mirada alerta, e inclusión del cuarto dedo en ambas manos (manos empuñadas).
Con respecto a sus pautas de desarrollo, no mantenía sostén cefálico y sus reflejos arcaicos estaban severamente comprometidos, con prensión palmar y plantar ausentes, reflejo Moro ausente y ángulo poplíteo de 130°.
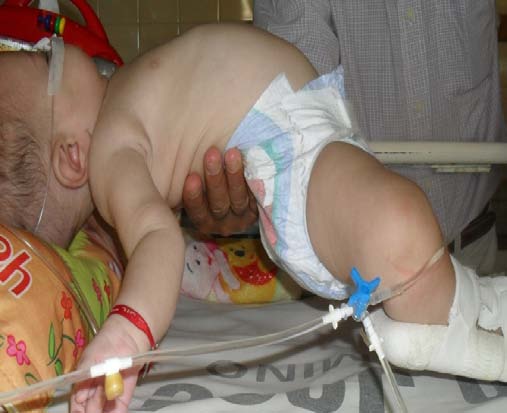
Se realizaron maniobras para evaluar su tono muscular, tales como la suspensión vertical, en la cuál el lactante parecía deslizarse entre las manos del explorador y la suspensión horizontal (ver foto), que demostraba marcada hipotonía de la raíz de los cuatro miembros. Escasa actividad muscular espontánea, con respuesta a estímulos, con movimientos a favor de la gravedad.
Se replanteó al paciente como Lactante Hipotónico.Planteos diagnósticos
El tono muscular se define como la resistencia al movimiento pasivo. La hipotonía es un síntoma común de disfunción neurológica que ocurre en enfermedades del encéfalo, la médula espinal, nervios y músculos. En la lactancia, los trastornos cerebrales superan en gran medida a los trastornos de la unidad motora.
El lactante con tono disminuido siempre constituye un reto diagnóstico. El adecuado enfoque diagnóstico de este síntoma, permitirá detectar enfermedades graves, potencialmente mortales. En el siguiente cuadro, podemos observar una clasificación topográfica de las causas más frecuentes de hipotonía, que resulta útil para su enfoque diagnóstico.
Muchas de las entidades expuestas no son evidentes en el período neonatal inmediato y llegan a la valoración neuropediátrica ya avanzada la lactancia, caso frecuente en la atrofia espinal, tanto en la forma grave Werdnig Hoffman, como en las formas inmediatas.
Respecto al paciente en cuestión, se llevó a cabo un plan de estudios, para llegar a la causa de su hipotonía. Los estudios realizados arrojaron los siguientes resultados:
• Dosaje TSH 0,430 (0,48-4,6) T4 1,3 ( VN 0,7-1,85)
• Citoquímico LCR: aspecto límpido, leucocitos 1, proteínas 0,46,
• Hemocultivos, Urocultivo, Cultivo LCR negativos
• Ecografía transfontanelar: no se observa dilatación ventricular.
• TAC cráneo: estructuras conservadas, sin alteraciones patológicas.
• TAC tórax: estructuras conservadas, sin alteraciones.
Dada la clínica del paciente, con datos compatibles con atrofia medular espinal, se decidió su derivación a centro de mayor complejidad para continuar con los estudios pertinentes, entre ellos estudios genéticos.
Una vez resuelta su dificultad respiratoria, fue derivado al Hospital de Niños P. Garrahan donde se lo evaluó multidisciplinariamente. Finalmente, los estudios genéticos confirmaron la sospecha diagnóstica de Atrofia de Werdnig Hoffmann.
Una vez dado de alta del Hospital P. Garrahan con indicaciones terapéuticas específicas, el paciente retornó a su hogar durante un mes, reingresando a la Unidad de Cuidados Intensivos de nuestro hospital por paro respiratorio, que fue resuelto con medidas de reanimación. Permaneció con dependencia de oxígeno, debido a la hipotonía marcada de los músculos de su caja torácica, alimentándose por SNG, por presentar dificultad en la deglución.
Lenta y progresivamente, la hipotonía continuó aumentando, siendo necesaria la ventilación no invasiva para lograr saturaciones de oxígeno adecuadas.
El paciente falleció a los seis meses de vida por claudicación respiratoria.
Comentarios
Las atrofias espinales son trastornos hereditarios de las neuronas motoras espinales y bulbares que causan atrofia y debilidad musculares, de forma simétrica y proximal, con predominio de extremidades inferiores, siendo respetadas las estructuras faciales y el intelecto.
La etiología es genética, monogénica, autosómica recesiva. La alteración más frecuente es la deleción del gen SMN, siempre en situación de homocigosidad, es decir, doble copia alterada. El mecanismo probable radica en un defecto en la muerte celular programada, de forma que la eliminación de las células, proceso normal durante la gestación, continúa tras el nacimiento. Al examen físico son característicos:
• Postura: brazos extendidos junto al tronco, con ligera pronación del
• Manos con flexión del tercer y cuarto dedo.
• Piernas abiertas y pegadas al plano de apoyo, con rodillas
• Abolición de reflejos de estiramiento.
• Respiración diafragmática, tórax en campana, infecciones
Existen tres variedades clínico - evolutivas:
Atrofia Werdnig Hoffmann o tipo I
El cuadro clínico puede comenzar desde el nacimiento hasta los 6 meses. Los recién nacidos afectados tienen debilidad generalizada, que afecta más los músculos proximales que los distales, hipotonía y arreflexia. La mayoría respira adecuadamente al principio y parecen alertas a pesar de la marcada debilidad.
Cuando la debilidad comienza en la lactancia, el deterioro de la fuerza puede ser brusco o progresivo. En ocasiones el lactante impresiona cierta mejoría, debido a que su desarrollo cerebral es normal, pero la progresión de la debilidad es implacable. No se logra la sedestación. Cuando se pierde el reflejo nauseoso, la alimentación se dificulta, la muerte se produce por aspiración y neumonía. Fallecen antes de los dos años de edad.
Existe una variante, la AME infantil con dificultad respiratoria tipo I, cuyo diagnóstico debe ser sospechado ante aquellos lactantes con dificultad respiratoria secundaria a debilidad diafragmática, llanto débil y paresia distal progresiva de miembros inferiores. En ausencia de soporte respiratorio, se produce la muerte súbita del lactante.
- Fallecimiento en la adolescencia. - No se logra la deambulación autónoma
- Fallecimiento en la edad adulta. - Se logra la deambulación
Para el diagnóstico de certeza de las atrofias espinales, se dispone de prueba genética molecular para el gen SMN. Alrededor del 95% de los individuos con AME, son homocigotos para la ausencia de los exones 7 y 8 del SMN.
La concentración sérica de CPK suele ser normal, si bien puede estar ligeramente elevada en los casos de rápida progresión de la debilidad muscular. El EMG muestra fibrilaciones y fasciculaciones en reposo, la velocidad de conducción de los nervios motores está conservada.
La biopsia muscular es innecesaria, dada la disponibilidad de pruebas genéticas. Los hallazgos anatomopatológicos en el músculo esquelético son característicos, muestran grupos de fibras pequeñas adyacentes a grupos de fibras de tamaño normal o hipertrofiado. Los principales diagnósticos diferenciales se realizan con cuadros clínicos no espinales, como las miopatías congénitas; la diferenciación es electrofisiológica.
El tratamiento es de sostén, por no disponerse de tratamiento eficaz para esta entidad. El consejo genético, en ausencia de datos pronósticos específicos, es la abstención reproductiva.
Dra. Varela, Constanza R.
SAMENVATTING VAN DE PRODUCTKENMERKEN NAAM VAN HET DIERGENEESMIDDEL Prascend 1 mg tabletten voor paarden 2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING Per tablet: Werkzaam bestanddeel: 1,0 mg pergolide (als pergolidemesilaat 1,31 mg) Hulpstoffen: Zie rubriek 6.1 voor een volledige lijst van hulpstoffen. 3. FARMACEUTISCHE VORM Tablet Roze, rechthoekige t
HEALTH HISTORY FORM FOR OFFICE FOR CHILDREN, YOUTH AND ADULTS PARTICIPATING IN NLOM CAMP PROGRAMS DUE 2 WEEKS PRIOR TO CAMP PLEASE PRINT OR TYPE The following information must be filled out by the parent/legal guardian or adult camper or staff member. The intent of this information is to provide camp health care personnel the background to provide appropriate care. We suggest yo
 Residente de tercer año de Pediatría, HIGA San José de Pergamino
Lactante de sexo masculino de dos meses de edad, que ingresa a nuestra sala de Pediatría con diagnóstico de atelectasia masiva. Refiere cuadro de 72 hs de evolución caracterizado por dificultad respiratoria e hiporexia.
Antecedentes personales: fruto de un embarazo controlado, G1 P1, parto eutócico, RNT de 40 semanas de gestación, peso: 3200 gr., score de Apgar vigoroso, internación conjunta. Caída tardía del cordón umbilical -a los 24 días de vida-. Lactancia materna exclusiva, con esquema de vacunación acorde a la edad.
Antecedentes familiares: madre, 19 años, sana; padre 26 años, sano. No refieren antecedentes familiares de relevancia.
A su ingreso, el paciente se encontraba alerta, en regular estado general, afebril, hemodinámicamente compensado, taquipneico, taquicárdico, conectado con el medio. Su auscultación evidenciaba regular entrada de aire bilateral, con disminución del murmullo vesicular en hemitórax izquierdo, mecánica ventilatoria exigida, con tiraje subcostal e intercostal y patrón tusígeno débil. Saturación oxígeno 92%, aire ambiente.
Se realizaron estudios de laboratorio de rutina, que no arrojaron resultados patológicos y radiografía de tórax, donde se evidenciaba una radioopacidad en lóbulo superior izquierdo, que tracciona el mediastino hacia su lado, compatible con atelectasia. Se tomaron hemocultivos.
Se indicaron medidas de sostén, oxigenoterapia, SNG, salbutamol 200mcg cada 4 hs, hidrocortisona 18mg/k, dexametasona 0,6mg./Kg./día, ampicilina 200 mg./Kg./día, gentamicina 5 mg./kg./día y kinesioterapia respiratoria.
Si bien el paciente ingresó por dificultad respiratoria, ciertos datos de su examen neurológico llamaban la atención, siendo francamente patológicos. En decúbito dorsal, el lactante se encontraba con sus cuatro extremidades en
extensión e hipotónicas; con tortícolis izquierda, mirada alerta, e inclusión del cuarto dedo en ambas manos (manos empuñadas).
Con respecto a sus pautas de desarrollo, no mantenía sostén cefálico y sus reflejos arcaicos estaban severamente comprometidos, con prensión palmar y plantar ausentes, reflejo Moro ausente y ángulo poplíteo de 130°.
Se realizaron maniobras para evaluar su tono muscular, tales como la suspensión vertical, en la cuál el lactante parecía deslizarse entre las manos del explorador y la suspensión horizontal (ver foto), que demostraba marcada hipotonía de la raíz de los cuatro miembros. Escasa actividad muscular espontánea, con respuesta a estímulos, con movimientos a favor de la gravedad.
Se replanteó al paciente como Lactante Hipotónico.
Planteos diagnósticos
Residente de tercer año de Pediatría, HIGA San José de Pergamino
Lactante de sexo masculino de dos meses de edad, que ingresa a nuestra sala de Pediatría con diagnóstico de atelectasia masiva. Refiere cuadro de 72 hs de evolución caracterizado por dificultad respiratoria e hiporexia.
Antecedentes personales: fruto de un embarazo controlado, G1 P1, parto eutócico, RNT de 40 semanas de gestación, peso: 3200 gr., score de Apgar vigoroso, internación conjunta. Caída tardía del cordón umbilical -a los 24 días de vida-. Lactancia materna exclusiva, con esquema de vacunación acorde a la edad.
Antecedentes familiares: madre, 19 años, sana; padre 26 años, sano. No refieren antecedentes familiares de relevancia.
A su ingreso, el paciente se encontraba alerta, en regular estado general, afebril, hemodinámicamente compensado, taquipneico, taquicárdico, conectado con el medio. Su auscultación evidenciaba regular entrada de aire bilateral, con disminución del murmullo vesicular en hemitórax izquierdo, mecánica ventilatoria exigida, con tiraje subcostal e intercostal y patrón tusígeno débil. Saturación oxígeno 92%, aire ambiente.
Se realizaron estudios de laboratorio de rutina, que no arrojaron resultados patológicos y radiografía de tórax, donde se evidenciaba una radioopacidad en lóbulo superior izquierdo, que tracciona el mediastino hacia su lado, compatible con atelectasia. Se tomaron hemocultivos.
Se indicaron medidas de sostén, oxigenoterapia, SNG, salbutamol 200mcg cada 4 hs, hidrocortisona 18mg/k, dexametasona 0,6mg./Kg./día, ampicilina 200 mg./Kg./día, gentamicina 5 mg./kg./día y kinesioterapia respiratoria.
Si bien el paciente ingresó por dificultad respiratoria, ciertos datos de su examen neurológico llamaban la atención, siendo francamente patológicos. En decúbito dorsal, el lactante se encontraba con sus cuatro extremidades en
extensión e hipotónicas; con tortícolis izquierda, mirada alerta, e inclusión del cuarto dedo en ambas manos (manos empuñadas).
Con respecto a sus pautas de desarrollo, no mantenía sostén cefálico y sus reflejos arcaicos estaban severamente comprometidos, con prensión palmar y plantar ausentes, reflejo Moro ausente y ángulo poplíteo de 130°.
Se realizaron maniobras para evaluar su tono muscular, tales como la suspensión vertical, en la cuál el lactante parecía deslizarse entre las manos del explorador y la suspensión horizontal (ver foto), que demostraba marcada hipotonía de la raíz de los cuatro miembros. Escasa actividad muscular espontánea, con respuesta a estímulos, con movimientos a favor de la gravedad.
Se replanteó al paciente como Lactante Hipotónico.
Planteos diagnósticos  Respecto al paciente en cuestión, se llevó a cabo un plan de estudios, para llegar a la causa de su hipotonía. Los estudios realizados arrojaron los siguientes resultados:
• Dosaje TSH 0,430 (0,48-4,6) T4 1,3 ( VN 0,7-1,85)
• Citoquímico LCR: aspecto límpido, leucocitos 1, proteínas 0,46,
• Hemocultivos, Urocultivo, Cultivo LCR negativos
• Ecografía transfontanelar: no se observa dilatación ventricular.
• TAC cráneo: estructuras conservadas, sin alteraciones patológicas.
• TAC tórax: estructuras conservadas, sin alteraciones.
Dada la clínica del paciente, con datos compatibles con atrofia medular espinal, se decidió su derivación a centro de mayor complejidad para continuar con los estudios pertinentes, entre ellos estudios genéticos.
Una vez resuelta su dificultad respiratoria, fue derivado al Hospital de Niños P. Garrahan donde se lo evaluó multidisciplinariamente. Finalmente, los estudios genéticos confirmaron la sospecha diagnóstica de Atrofia de Werdnig Hoffmann.
Una vez dado de alta del Hospital P. Garrahan con indicaciones terapéuticas específicas, el paciente retornó a su hogar durante un mes, reingresando a la Unidad de Cuidados Intensivos de nuestro hospital por paro respiratorio, que fue resuelto con medidas de reanimación. Permaneció con dependencia de oxígeno, debido a la hipotonía marcada de los músculos de su caja torácica, alimentándose por SNG, por presentar dificultad en la deglución.
Lenta y progresivamente, la hipotonía continuó aumentando, siendo necesaria la ventilación no invasiva para lograr saturaciones de oxígeno adecuadas.
El paciente falleció a los seis meses de vida por claudicación respiratoria.
Comentarios
Respecto al paciente en cuestión, se llevó a cabo un plan de estudios, para llegar a la causa de su hipotonía. Los estudios realizados arrojaron los siguientes resultados:
• Dosaje TSH 0,430 (0,48-4,6) T4 1,3 ( VN 0,7-1,85)
• Citoquímico LCR: aspecto límpido, leucocitos 1, proteínas 0,46,
• Hemocultivos, Urocultivo, Cultivo LCR negativos
• Ecografía transfontanelar: no se observa dilatación ventricular.
• TAC cráneo: estructuras conservadas, sin alteraciones patológicas.
• TAC tórax: estructuras conservadas, sin alteraciones.
Dada la clínica del paciente, con datos compatibles con atrofia medular espinal, se decidió su derivación a centro de mayor complejidad para continuar con los estudios pertinentes, entre ellos estudios genéticos.
Una vez resuelta su dificultad respiratoria, fue derivado al Hospital de Niños P. Garrahan donde se lo evaluó multidisciplinariamente. Finalmente, los estudios genéticos confirmaron la sospecha diagnóstica de Atrofia de Werdnig Hoffmann.
Una vez dado de alta del Hospital P. Garrahan con indicaciones terapéuticas específicas, el paciente retornó a su hogar durante un mes, reingresando a la Unidad de Cuidados Intensivos de nuestro hospital por paro respiratorio, que fue resuelto con medidas de reanimación. Permaneció con dependencia de oxígeno, debido a la hipotonía marcada de los músculos de su caja torácica, alimentándose por SNG, por presentar dificultad en la deglución.
Lenta y progresivamente, la hipotonía continuó aumentando, siendo necesaria la ventilación no invasiva para lograr saturaciones de oxígeno adecuadas.
El paciente falleció a los seis meses de vida por claudicación respiratoria.
Comentarios